Myer Phillip R, Kim MinSeok, Freetly Harvey C, Smith Timothy P L
Department of Animal Science, University of Tennessee Institute of Agriculture, University of Tennessee, Knoxville, TN 37996, USA.
USDA-ARS, U.S. Meat Animal Research Center, Clay Center NE 68933 , USA.
Data Brief. 2016 Jul 19;8:1048-53. doi: 10.1016/j.dib.2016.07.027. eCollection 2016 Sep.
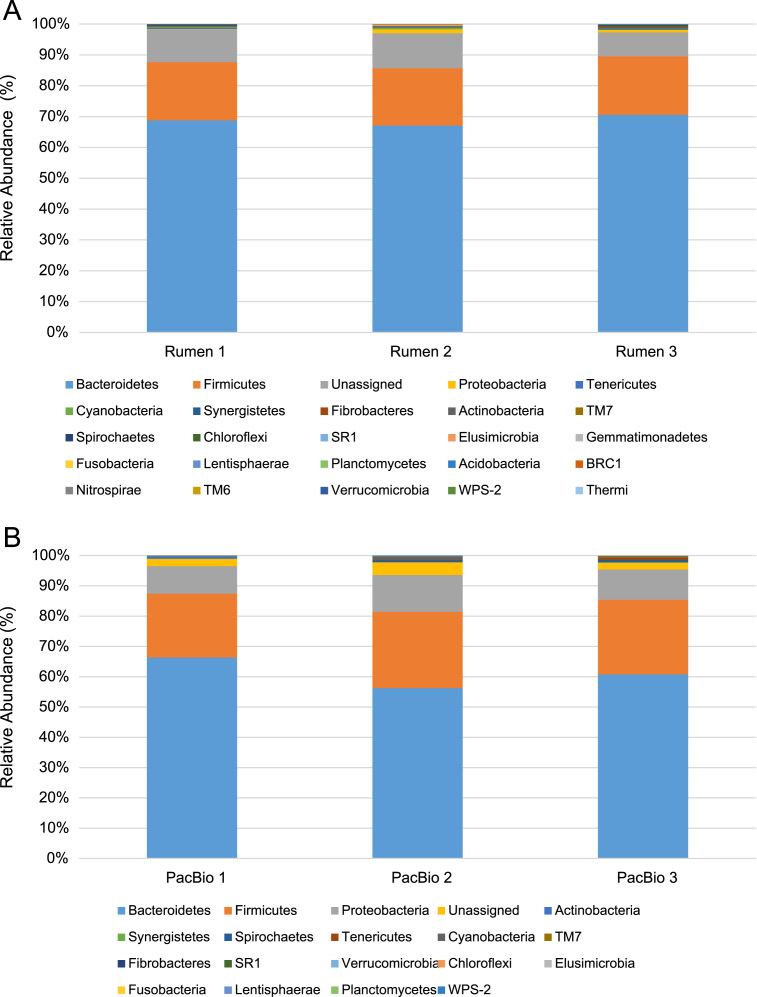
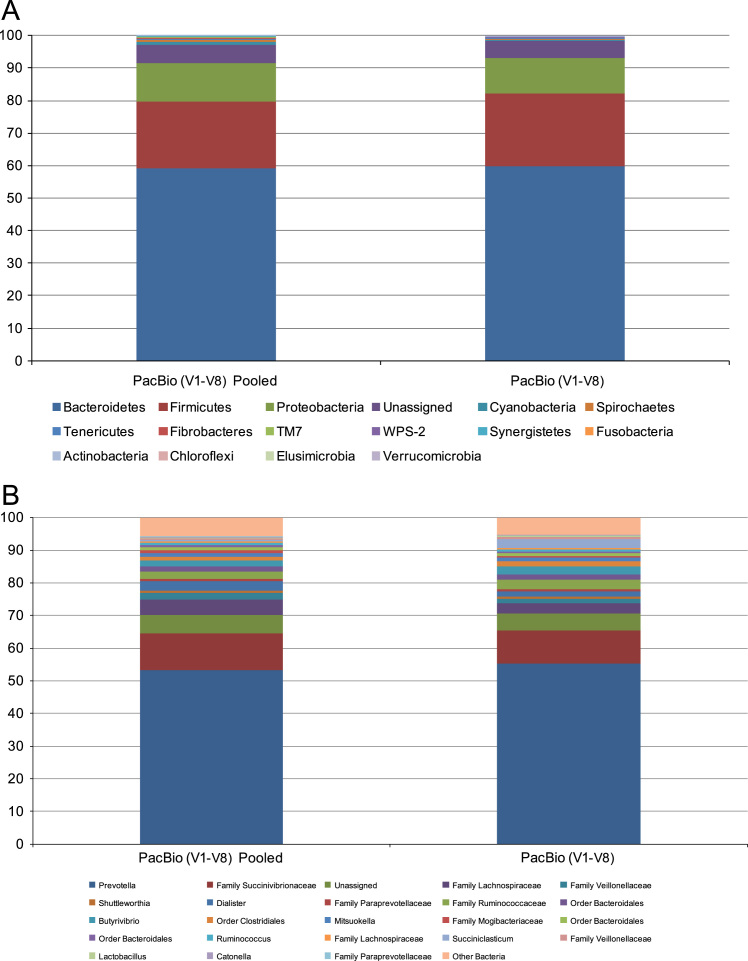
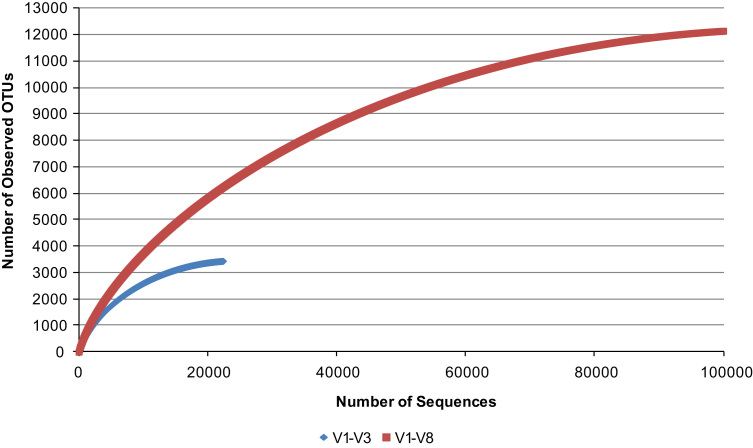
Amplicon sequencing utilizing next-generation platforms has significantly transformed how research is conducted, specifically microbial ecology. However, primer and sequencing platform biases can confound or change the way scientists interpret these data. The Pacific Biosciences RSII instrument may also preferentially load smaller fragments, which may also be a function of PCR product exhaustion during sequencing. To further examine theses biases, data is provided from 16S rRNA rumen community analyses. Specifically, data from the relative phylum-level abundances for the ruminal bacterial community are provided to determine between-sample variability. Direct sequencing of metagenomic DNA was conducted to circumvent primer-associated biases in 16S rRNA reads and rarefaction curves were generated to demonstrate adequate coverage of each amplicon. PCR products were also subjected to reduced amplification and pooling to reduce the likelihood of PCR product exhaustion during sequencing on the Pacific Biosciences platform. The taxonomic profiles for the relative phylum-level and genus-level abundance of rumen microbiota as a function of PCR pooling for sequencing on the Pacific Biosciences RSII platform were provided. For more information, see "Evaluation of 16S rRNA amplicon sequencing using two next-generation sequencing technologies for phylogenetic analysis of the rumen bacterial community in steers" P.R. Myer, M. Kim, H.C. Freetly, T.P.L. Smith (2016) [1].
利用新一代平台进行的扩增子测序显著改变了研究的开展方式,尤其是微生物生态学研究。然而,引物和测序平台偏差可能会混淆或改变科学家解读这些数据的方式。太平洋生物科学公司的RSII仪器可能还会优先加载较小的片段,这也可能是测序过程中PCR产物耗尽的一种表现。为了进一步研究这些偏差,我们提供了来自16S rRNA瘤胃群落分析的数据。具体而言,提供了瘤胃细菌群落相对门水平丰度的数据,以确定样本间的变异性。对宏基因组DNA进行直接测序,以规避16S rRNA读数中与引物相关的偏差,并生成稀疏曲线以证明每个扩增子的覆盖范围足够。PCR产物还进行了减少扩增和合并,以降低在太平洋生物科学平台上测序时PCR产物耗尽的可能性。提供了瘤胃微生物群在太平洋生物科学RSII平台上测序时,相对门水平和属水平丰度的分类学概况与PCR合并的函数关系。更多信息,请参阅“使用两种新一代测序技术对育肥牛瘤胃细菌群落进行系统发育分析的16S rRNA扩增子测序评估”,P.R.迈尔、M.金、H.C.弗里特利、T.P.L.史密斯(2016年)[1]。