Utsunomiya Adam T H, Santos Daniel J A, Boison Solomon A, Utsunomiya Yuri T, Milanesi Marco, Bickhart Derek M, Ajmone-Marsan Paolo, Sölkner Johann, Garcia José F, da Fonseca Ricardo, da Silva Marcos V G B
Faculdade de Ciências Agrárias e Veterinárias, Universidade Estadual Paulista - UNESP, Campus de Jaboticabal, São Paulo, Brasil.
Nofima, Ås, Norway.
BMC Genomics. 2016 Sep 5;17(1):705. doi: 10.1186/s12864-016-3049-8.
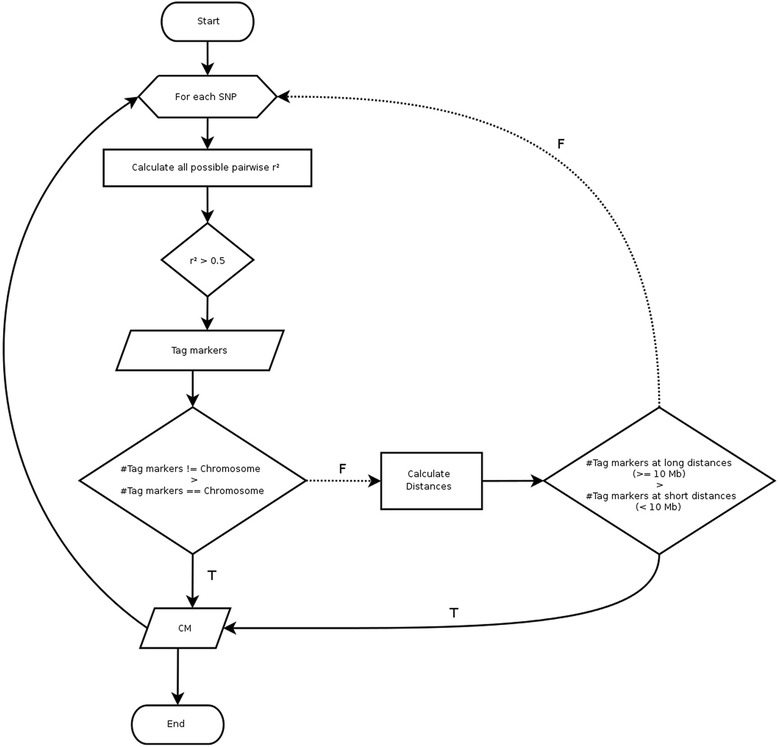
Misassembly signatures, created by shuffling the order of sequences while assembling a genome, can be detected by the unexpected behavior of marker linkage disequilibrium (LD) decay. We developed a heuristic process to identify misassembly signatures, applied it to the bovine reference genome assembly (UMDv3.1) and presented the consequences of misassemblies in two case studies.
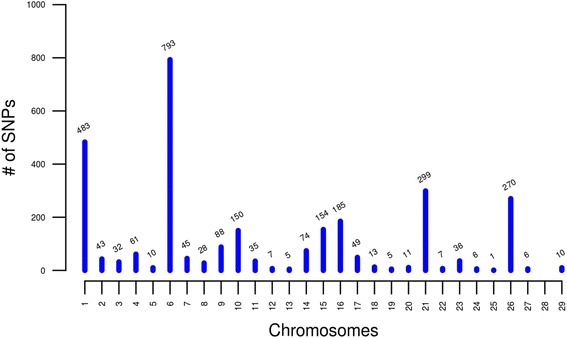
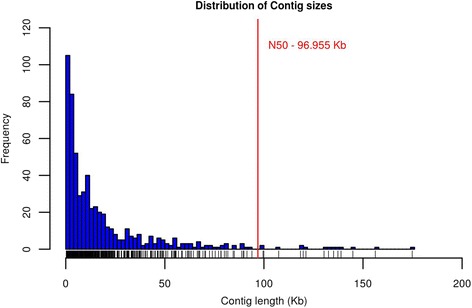
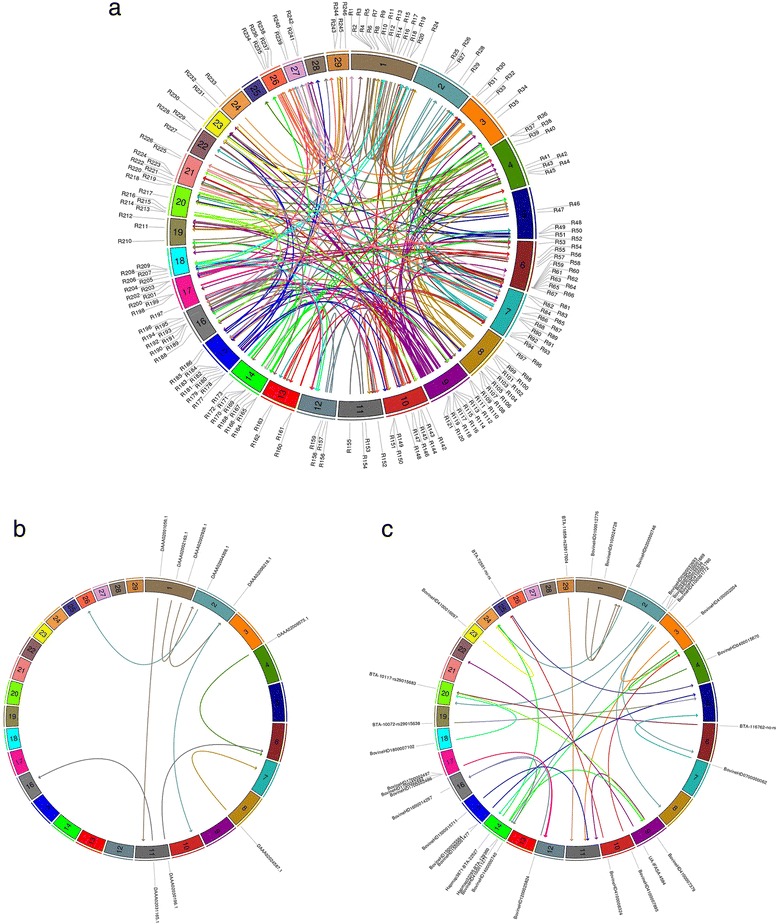
We identified 2,906 single nucleotide polymorphism (SNP) markers presenting unexpected LD decay behavior in 626 putative misassembled contigs, which comprised less than 1 % of the whole genome. Although this represents a small fraction of the reference sequence, these poorly assembled segments can lead to severe implications to local genome context. For instance, we showed that one of the misassembled regions mapped to the POLL locus, which affected the annotation of positional candidate genes in a GWAS case study for polledness in Nellore (Bos indicus beef cattle). Additionally, we found that poorly performing markers in imputation mapped to putative misassembled regions, and that correction of marker positions based on LD was capable to recover imputation accuracy.
This heuristic approach can be useful to cross validate reference assemblies and to filter out markers located at low confidence genomic regions before conducting downstream analyses.
在组装基因组时通过打乱序列顺序产生的错误组装特征,可通过标记连锁不平衡(LD)衰减的异常行为来检测。我们开发了一种启发式方法来识别错误组装特征,并将其应用于牛参考基因组组装(UMDv3.1),并在两个案例研究中展示了错误组装的后果。
我们在626个假定错误组装的重叠群中鉴定出2906个呈现异常LD衰减行为的单核苷酸多态性(SNP)标记,这些重叠群占整个基因组的比例不到1%。尽管这仅占参考序列的一小部分,但这些组装不佳的片段可能会对局部基因组背景产生严重影响。例如,我们发现其中一个错误组装区域映射到POLL基因座,这在一项针对内洛尔牛(印度瘤牛)无角性状的全基因组关联研究案例中影响了位置候选基因的注释。此外,我们发现插补效果不佳的标记映射到假定的错误组装区域,并且基于LD校正标记位置能够恢复插补准确性。
这种启发式方法可用于交叉验证参考组装,并在进行下游分析之前筛选出位于低置信度基因组区域的标记。