Sun Zewei, Zhou Dongchen, Xie Xudong, Wang Shuai, Wang Zhen, Zhao Wenting, Xu Hongfei, Zheng Liangrong
Department of Cardiology, The First Affiliated Hospital, College of Medicine, Zhejiang University, No. 79 Qingchun Road, Hangzhou, 310003, China.
Department of Cardiothoracic Surgery, The First Affiliated Hospital, College of Medicine, Zhejiang University, No.79 Qingchun Road, Hangzhou, 310003, China.
Basic Res Cardiol. 2016 Nov;111(6):63. doi: 10.1007/s00395-016-0584-z. Epub 2016 Sep 22.
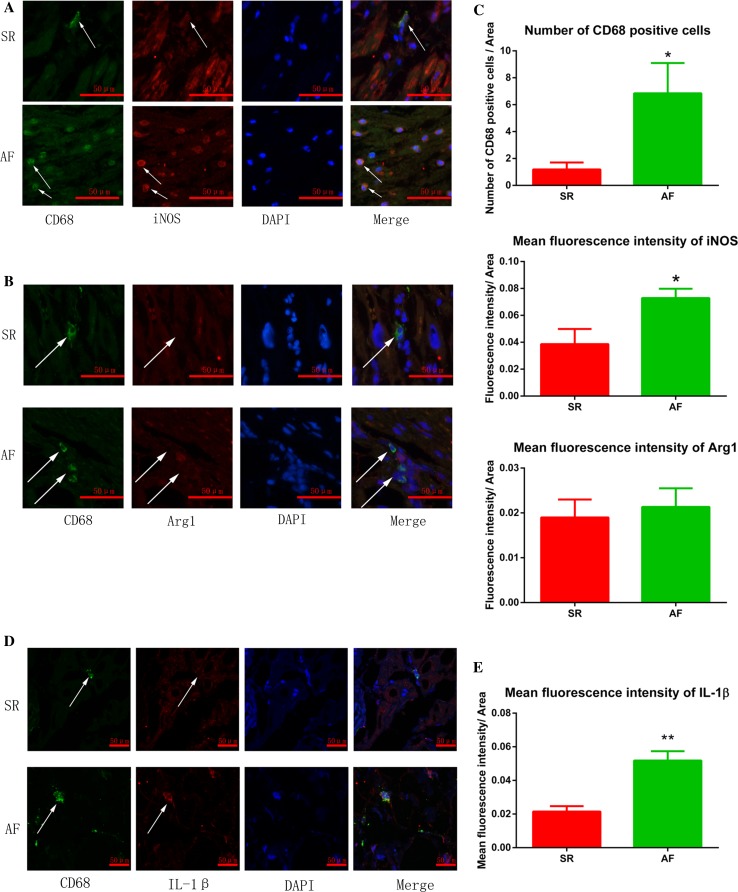
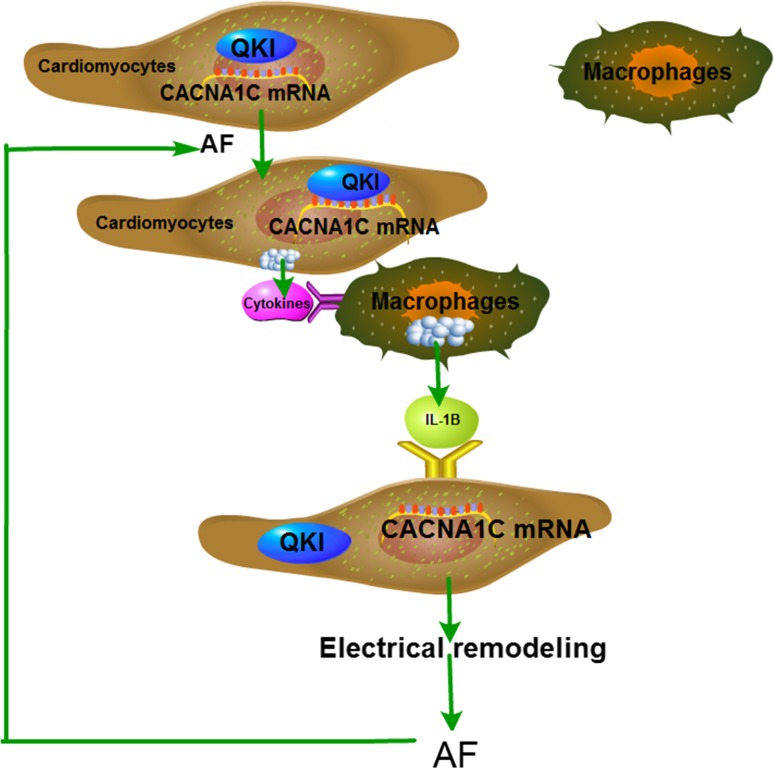
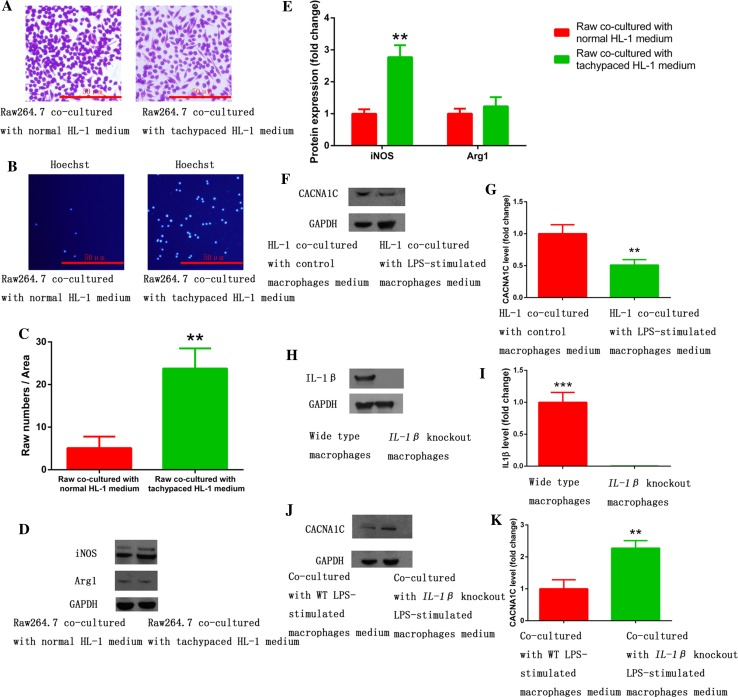
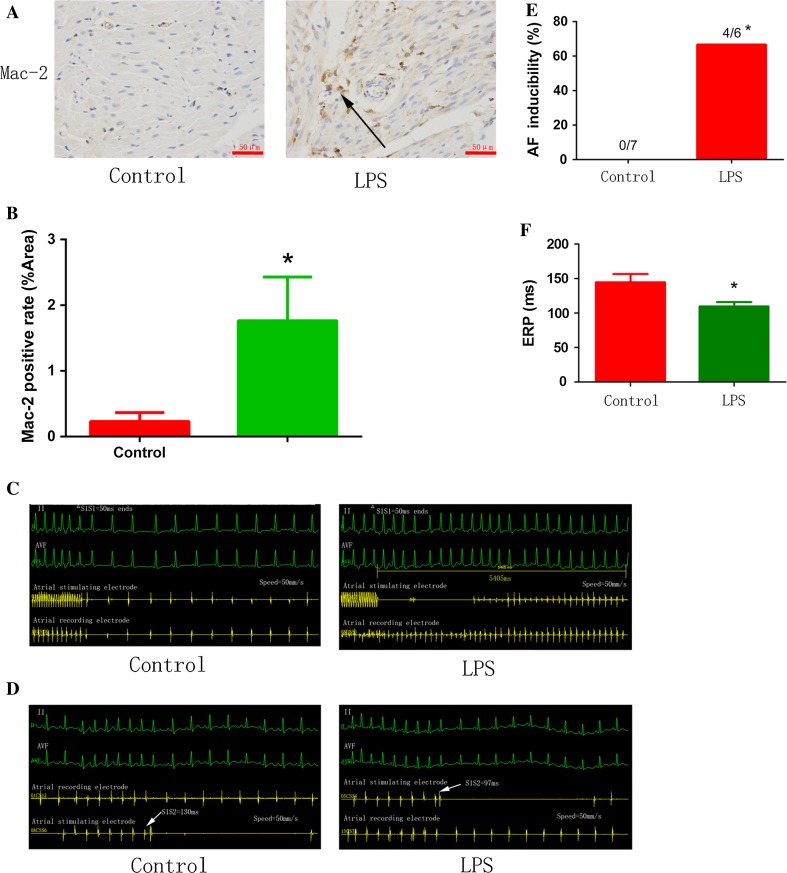
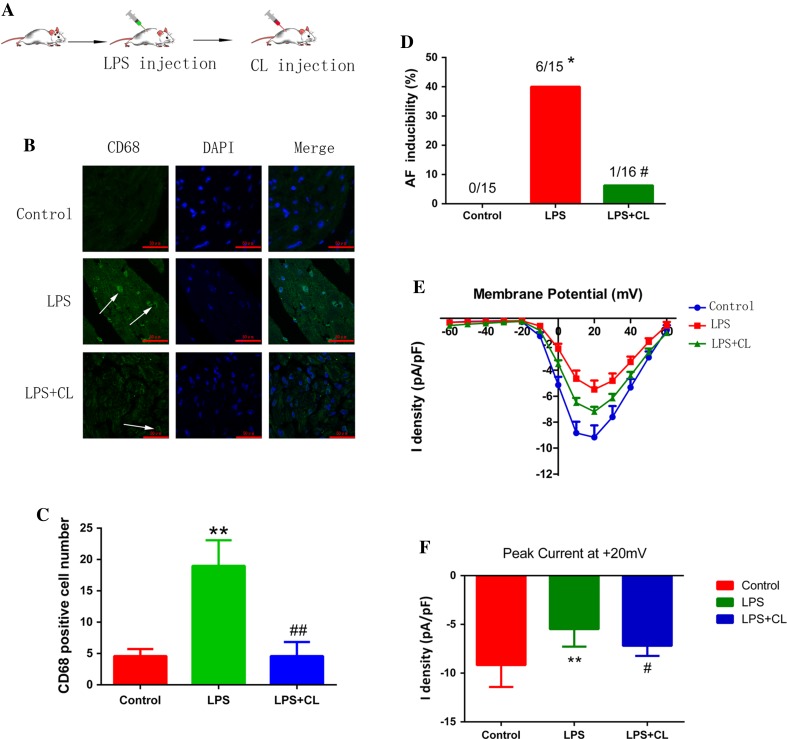
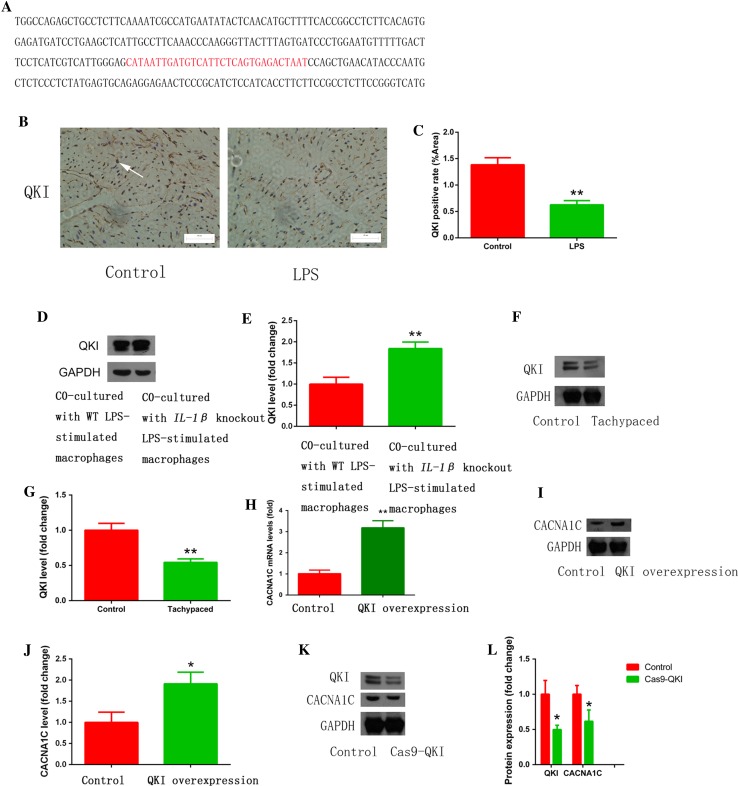
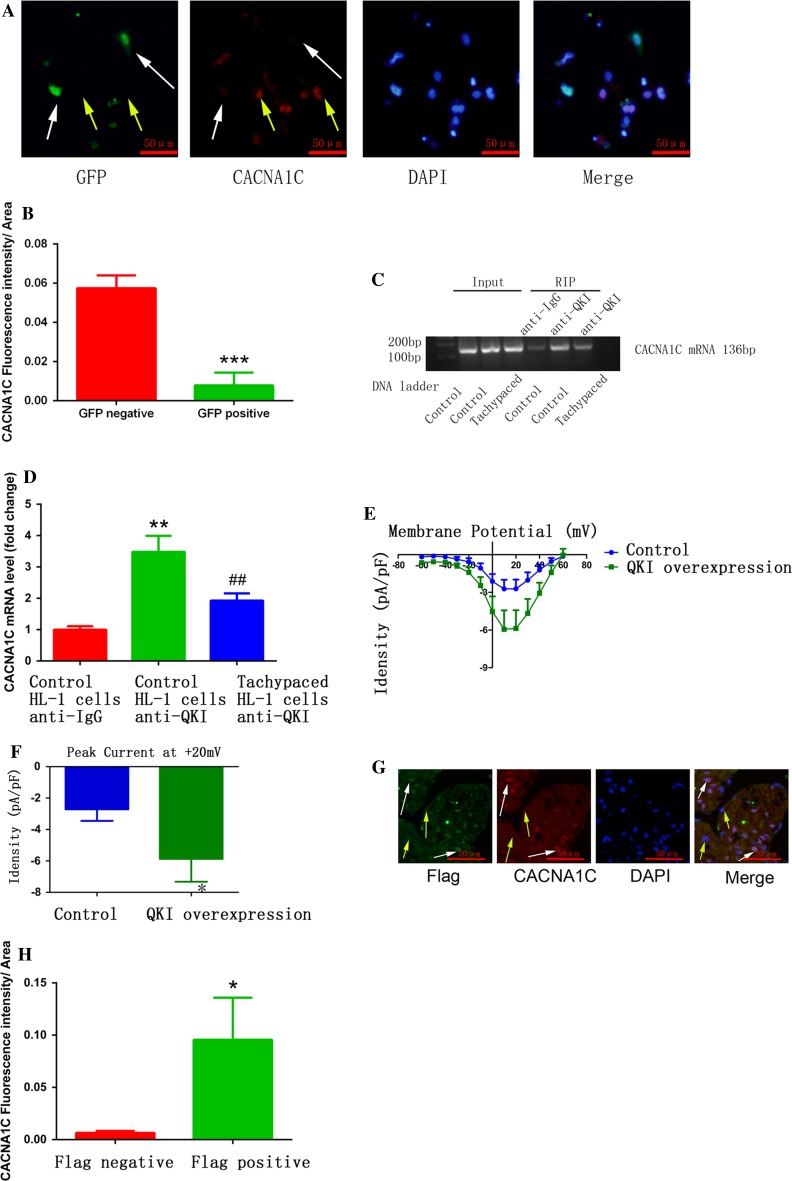
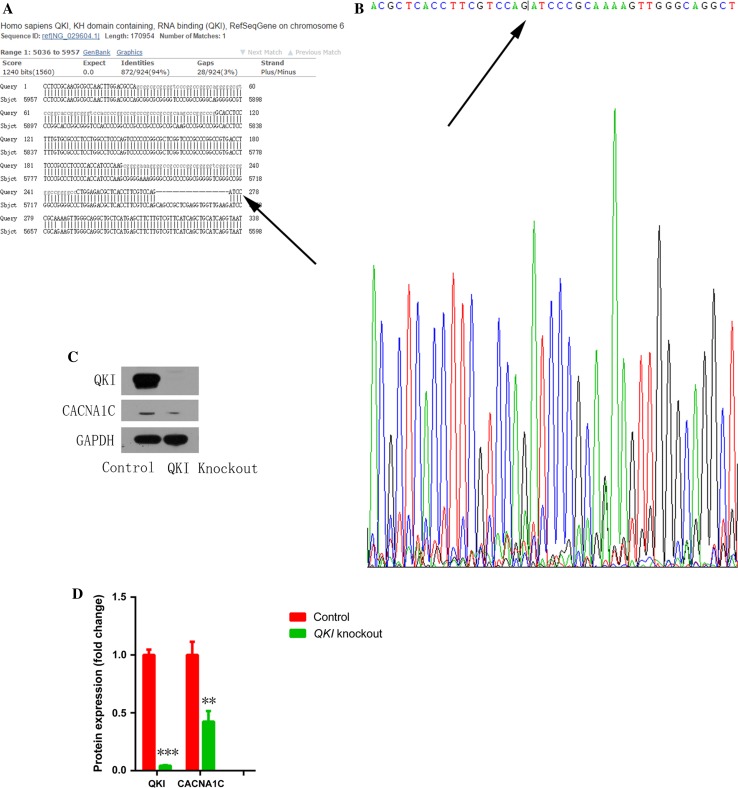
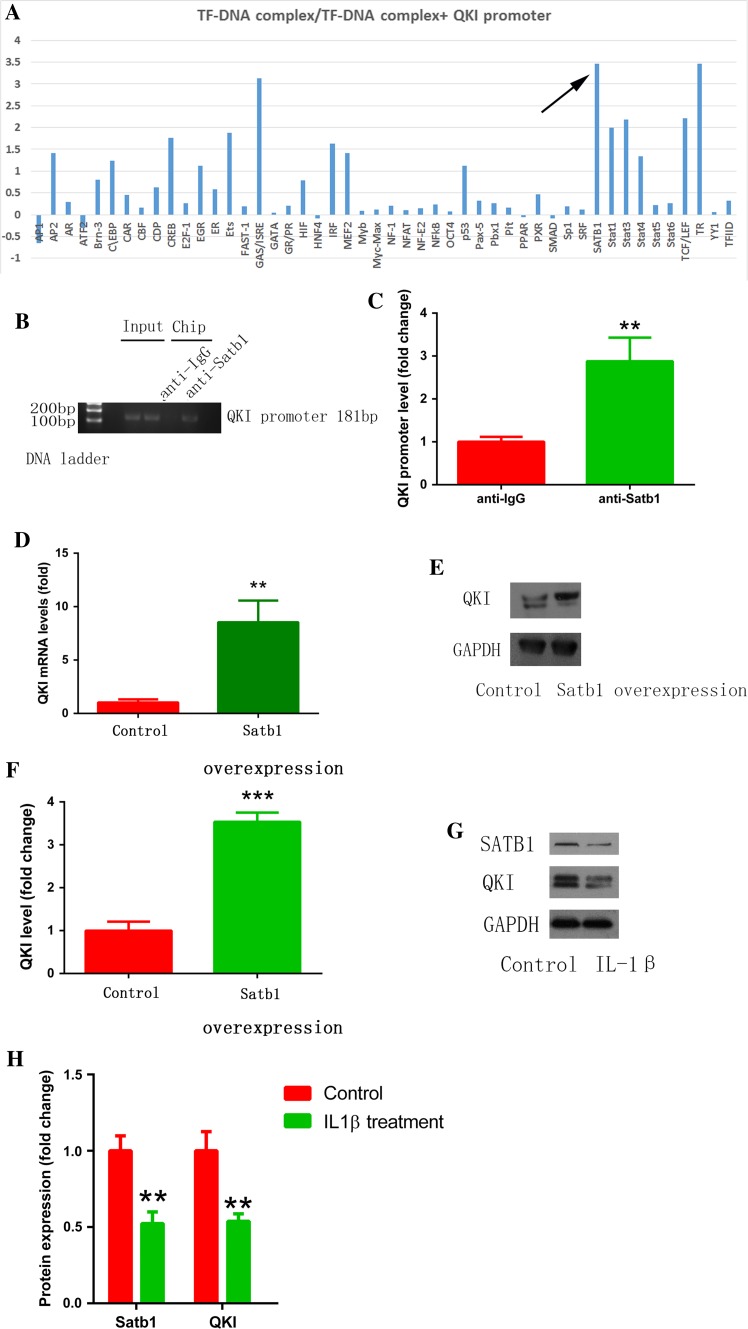
Increased macrophage accumulation occurs in the atria of patients with atrial fibrillation (AF). However, the phenotype and functions of the macrophages in AF remain unclear. We investigated the macrophage-atrial myocyte interaction in AF patients and found that the increased macrophages were mainly pro-inflammatory macrophages (iNOS, Arg1). Tachypacing of HL-1 atrial myocytes also led to pro-inflammatory macrophage polarization. In addition, lipopolysaccharide (LPS)-stimulated pro-inflammatory macrophages-induced atrial electrical remodeling, evidenced by increased AF incidence and decreased atrial effective refractory period and L-type calcium currents (I ) in both canine and mouse AF models. Depletion of macrophages relieved LPS-induced atrial electrical remodeling, confirming the role of pro-inflammatory macrophages in the pathogenesis of AF. We also found that the effect of LPS-stimulated macrophages on atrial myocytes was mediated by secretion of interleukin 1 beta (IL-1β), which inhibited atrial myocyte quaking protein (QKI) expression. IL-1β knockout in macrophages restored the LPS-stimulated macrophage-induced inhibition of QKI and CACNA1C (α1C subunit of L-type calcium channel) in atrial myocytes. Meanwhile, QKI overexpression in atrial myocytes restored the LPS-stimulated macrophage-induced electrical remodeling through enhanced binding of QKI to CACNA1C mRNA, which upregulated the expression of CACNA1C as well as I . In contrast, QKI knockout inhibited CACNA1C expression. Finally, using transcription factor activation profiling plate array and chromatin immunoprecipitation, we revealed that special AT-rich sequence binding protein 1 activated QKI transcription. Taken together, our study uncovered the functional interaction between macrophages and atrial myocytes in AF. AF induced pro-inflammatory macrophage polarization while pro-inflammatory macrophages exacerbated atrial electrical remodeling by secreting IL-1β, further inhibiting QKI expression in atrial myocytes, which contributed to I downregulation. Our study demonstrates a novel molecular mechanism underlying the pathogenesis and progression of AF and suggests that QKI is a potential therapeutic target.
巨噬细胞在房颤(AF)患者心房中的积聚增加。然而,AF中巨噬细胞的表型和功能仍不清楚。我们研究了AF患者中巨噬细胞与心房肌细胞的相互作用,发现增加的巨噬细胞主要是促炎性巨噬细胞(诱导型一氧化氮合酶、精氨酸酶1)。HL-1心房肌细胞的快速起搏也导致促炎性巨噬细胞极化。此外,脂多糖(LPS)刺激的促炎性巨噬细胞诱导心房电重构,在犬和小鼠AF模型中表现为AF发生率增加、心房有效不应期缩短以及L型钙电流(I )降低。巨噬细胞耗竭减轻了LPS诱导的心房电重构,证实了促炎性巨噬细胞在AF发病机制中的作用。我们还发现,LPS刺激的巨噬细胞对心房肌细胞的作用是通过白细胞介素1β(IL-1β)的分泌介导的,IL-1β抑制心房肌细胞震颤蛋白(QKI)的表达。巨噬细胞中的IL-1β基因敲除恢复了LPS刺激的巨噬细胞诱导的心房肌细胞中QKI和L型钙通道α1C亚基(CACNA1C)的抑制。同时,心房肌细胞中QKI的过表达通过增强QKI与CACNA1C mRNA的结合恢复了LPS刺激的巨噬细胞诱导的电重构,从而上调了CACNA1C以及I 的表达。相反,QKI基因敲除抑制了CACNA1C的表达。最后,使用转录因子激活分析板阵列和染色质免疫沉淀,我们发现富含AT序列结合蛋白1激活了QKI转录。综上所述,我们的研究揭示了AF中巨噬细胞与心房肌细胞之间的功能相互作用。AF诱导促炎性巨噬细胞极化,而促炎性巨噬细胞通过分泌IL-1β加剧心房电重构,进一步抑制心房肌细胞中QKI的表达,这导致I 下调。我们的研究揭示了AF发病机制和进展的一种新的分子机制,并表明QKI是一个潜在的治疗靶点。