Choi Yong Seon, de Mattos Ana Barbosa Marcondes, Shao Dan, Li Tao, Nabben Miranda, Kim Maengjo, Wang Wang, Tian Rong, Kolwicz Stephen C
Mitochondria and Metabolism Center, Department of Anesthesiology & Pain Medicine, University of Washington, Seattle, WA, United States.
Mitochondria and Metabolism Center, Department of Anesthesiology & Pain Medicine, University of Washington, Seattle, WA, United States.
J Mol Cell Cardiol. 2016 Nov;100:64-71. doi: 10.1016/j.yjmcc.2016.09.001. Epub 2016 Sep 28.
Diastolic dysfunction is a common feature in many heart failure patients with preserved ejection fraction and has been associated with altered myocardial metabolism in hypertensive and diabetic patients. Therefore, metabolic interventions to improve diastolic function are warranted. In mice with a germline cardiac-specific deletion of acetyl CoA carboxylase 2 (ACC2), systolic dysfunction induced by pressure-overload was prevented by maintaining cardiac fatty acid oxidation (FAO). However, it has not been evaluated whether this strategy would prevent the development of diastolic dysfunction in the adult heart.
To test the hypothesis that augmenting cardiac FAO is protective against angiotensin II (AngII)-induced diastolic dysfunction in an adult mouse heart.
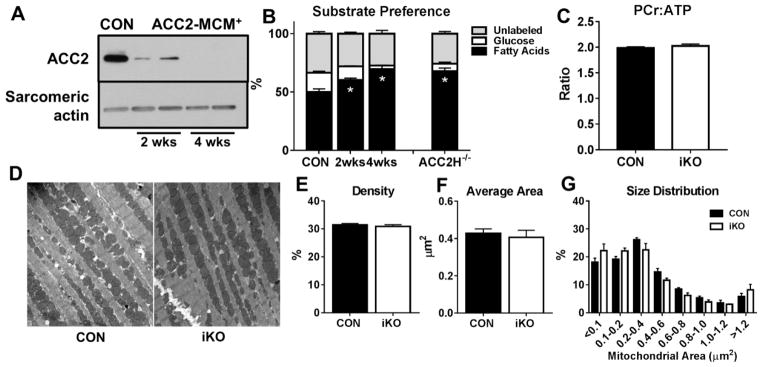
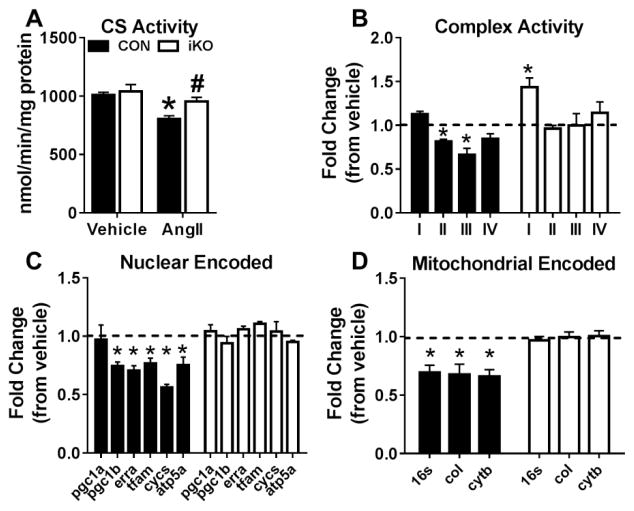
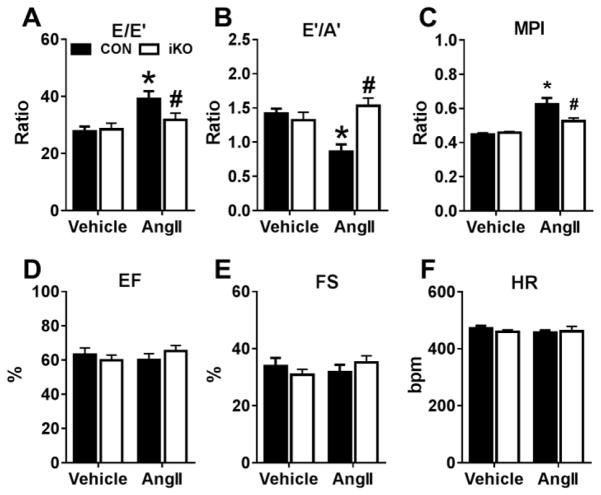
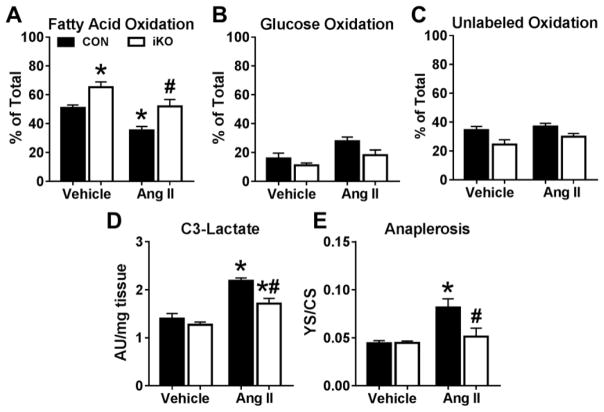
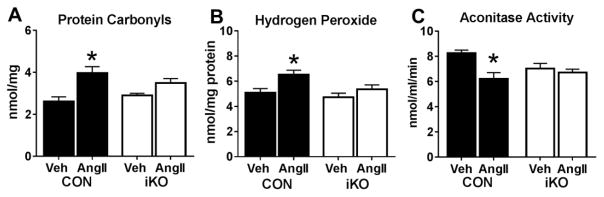
We generated a mouse model to induce cardiac-specific deletion of ACC2 in adult mice. Tamoxifen treatment (20mg/kg/day for 5days) was sufficient to delete ACC2 protein and increase cardiac FAO by 50% in ACC2 flox/flox-MerCreMer mice (iKO). After 4weeks of AngII (1.1mg/kg/day), delivered by osmotic mini-pumps, iKO mice showed normalized E/E' and E'/A' ratios compared to AngII treated controls (CON). The prevention of diastolic dysfunction in iKO-AngII was accompanied by maintained FAO and reduced glycolysis and anaplerosis. Furthermore, iKO-AngII hearts had a~50% attenuation of cardiac hypertrophy and fibrosis compared to CON. In addition, maintenance of FAO in iKO hearts suppressed AngII-associated increases in oxidative stress and sustained mitochondrial respiratory complex activities.
These data demonstrate that impaired FAO is a contributor to the development of diastolic dysfunction induced by AngII. Maintenance of FAO in this model leads to an attenuation of hypertrophy, reduces fibrosis, suppresses increases in oxidative stress, and maintains mitochondrial function. Therefore, targeting mitochondrial FAO is a promising therapeutic strategy for the treatment of diastolic dysfunction.
舒张功能障碍是许多射血分数保留的心力衰竭患者的常见特征,并且与高血压和糖尿病患者的心肌代谢改变有关。因此,有必要采取代谢干预措施来改善舒张功能。在心脏特异性乙酰辅酶A羧化酶2(ACC2)基因敲除的小鼠中,通过维持心脏脂肪酸氧化(FAO)可预防压力超负荷诱导的收缩功能障碍。然而,尚未评估该策略是否能预防成年心脏舒张功能障碍的发生。
验证增强心脏FAO对成年小鼠心脏中血管紧张素II(AngII)诱导的舒张功能障碍具有保护作用这一假说。
我们构建了一个在成年小鼠中诱导心脏特异性ACC2缺失的小鼠模型。在ACC2flox/flox-MerCreMer小鼠(iKO)中,给予他莫昔芬治疗(20mg/kg/天,持续5天)足以删除ACC2蛋白并使心脏FAO增加50%。在通过渗透微型泵给予AngII(1.1mg/kg/天)4周后,与AngII处理的对照组(CON)相比,iKO小鼠的E/E'和E'/A'比值恢复正常。iKO-AngII组舒张功能障碍的预防伴随着FAO的维持以及糖酵解和回补反应的减少。此外,与CON组相比,iKO-AngII组心脏的肥大和纤维化减轻了约50%。此外,iKO心脏中FAO的维持抑制了AngII相关的氧化应激增加,并维持了线粒体呼吸复合体的活性。
这些数据表明,FAO受损是AngII诱导的舒张功能障碍发生的一个因素。在该模型中维持FAO可减轻肥大、减少纤维化、抑制氧化应激增加并维持线粒体功能。因此,靶向线粒体FAO是治疗舒张功能障碍的一种有前景的治疗策略。