Hasegawa Eiko, Sawa Naoki, Hoshino Junichi, Suwabe Tatsuya, Hayami Noriko, Yamanouchi Masayuki, Sekine Akinari, Hiramatsu Rikako, Imafuku Aya, Kawada Masahiro, Ubara Yoshifumi, Imamura Tsunao, Takaichi Kenmei
Nephrology Center, Toranomon Hospital, Japan.
Intern Med. 2016;55(20):3009-3012. doi: 10.2169/internalmedicine.55.6818. Epub 2016 Oct 15.
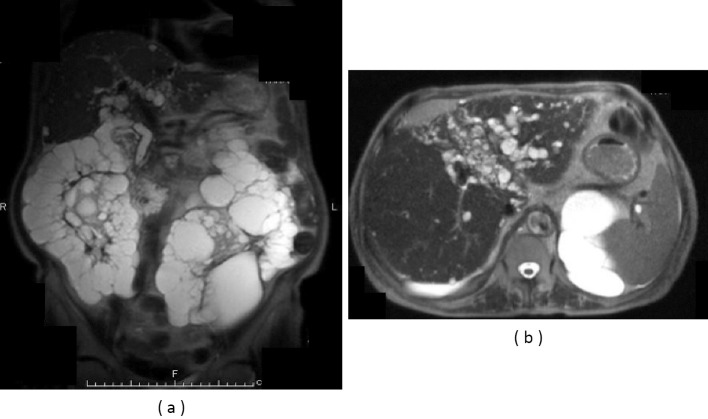
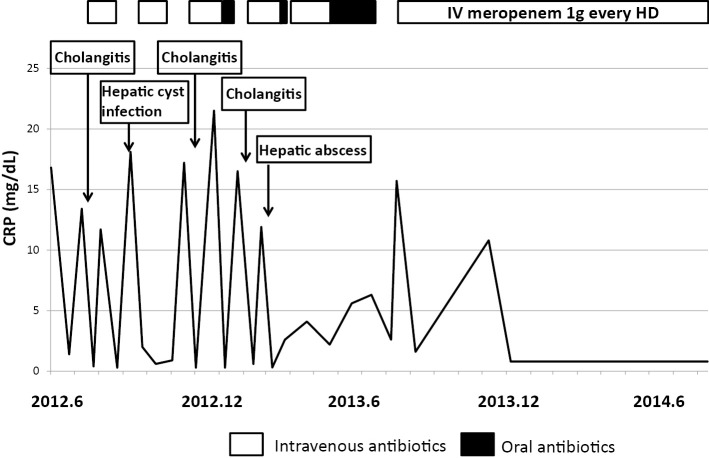
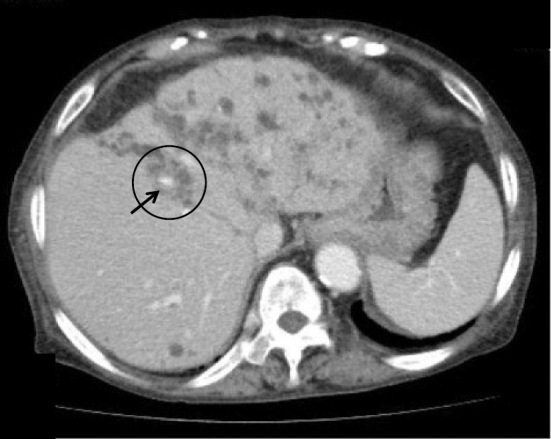
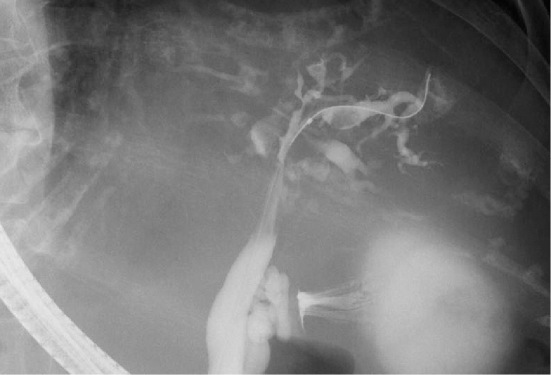
We herein present a rare case of an autosomal dominant polycystic kidney disease (ADPKD) patient with Caroli's disease, a congenital embryonic biliary tree ductal plate abnormality often associated with autosomal recessive polycystic kidney disease. A 76-year-old woman with ADPKD on hemodialysis was admitted to our hospital with recurrent cholangitis and hepatobiliary stones. Caroli's disease was diagnosed according to typical imaging findings of cystic intrahepatic bile duct dilatation and the central dot sign. Hepatobiliary system abnormalities such as Caroli's disease should be considered in febrile ADPKD patients, even in the absence of typical clinical signs or symptoms.
我们在此报告一例罕见的常染色体显性多囊肾病(ADPKD)患者合并卡罗里病,卡罗里病是一种先天性胚胎胆管板异常疾病,常与常染色体隐性多囊肾病相关。一名76岁接受血液透析的ADPKD女性患者因复发性胆管炎和肝胆结石入住我院。根据肝内胆管囊性扩张和中心点征的典型影像学表现诊断为卡罗里病。对于发热的ADPKD患者,即使没有典型的临床体征或症状,也应考虑卡罗里病等肝胆系统异常情况。