Shannon Casey P, Balshaw Robert, Chen Virginia, Hollander Zsuzsanna, Toma Mustafa, McManus Bruce M, FitzGerald J Mark, Sin Don D, Ng Raymond T, Tebbutt Scott J
PROOF Centre of Excellence, Vancouver, BC, Canada.
Centre for Heart Lung Innovation, University of British Columbia, Vancouver, BC, Canada.
BMC Genomics. 2017 Jan 6;18(1):43. doi: 10.1186/s12864-016-3460-1.
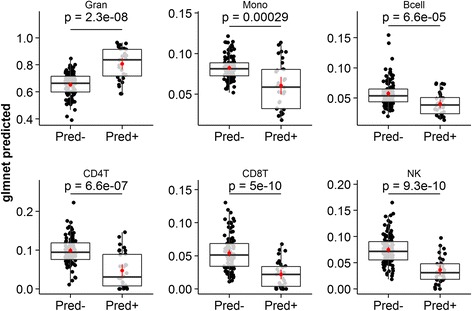
Measuring genome-wide changes in transcript abundance in circulating peripheral whole blood is a useful way to study disease pathobiology and may help elucidate the molecular mechanisms of disease, or discovery of useful disease biomarkers. The sensitivity and interpretability of analyses carried out in this complex tissue, however, are significantly affected by its dynamic cellular heterogeneity. It is therefore desirable to quantify this heterogeneity, either to account for it or to better model interactions that may be present between the abundance of certain transcripts, specific cell types and the indication under study. Accurate enumeration of the many component cell types that make up peripheral whole blood can further complicate the sample collection process, however, and result in additional costs. Many approaches have been developed to infer the composition of a sample from high-dimensional transcriptomic and, more recently, epigenetic data. These approaches rely on the availability of isolated expression profiles for the cell types to be enumerated. These profiles are platform-specific, suitable datasets are rare, and generating them is expensive. No such dataset exists on the Affymetrix Gene ST platform.
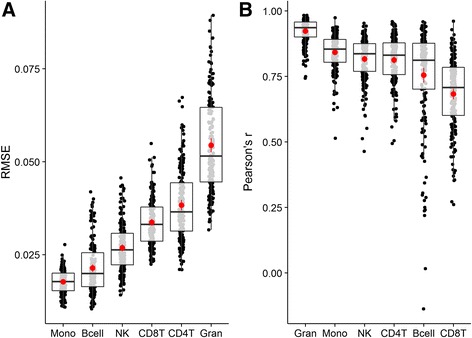
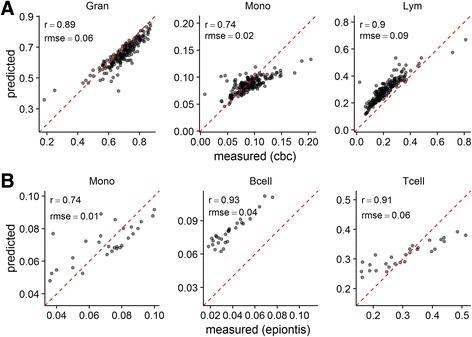
We present 'Enumerateblood', a freely-available and open source R package that exposes a multi-response Gaussian model capable of accurately predicting the composition of peripheral whole blood samples from Affymetrix Gene ST expression profiles, outperforming other current methods when applied to Gene ST data.
'Enumerateblood' significantly improves our ability to study disease pathobiology from whole blood gene expression assayed on the popular Affymetrix Gene ST platform by allowing a more complete study of the various components of this complex tissue without the need for additional data collection. Future use of the model may allow for novel insights to be generated from the ~400 Affymetrix Gene ST blood gene expression datasets currently available on the Gene Expression Omnibus (GEO) website.
测量循环外周全血中转录本丰度的全基因组变化是研究疾病病理生物学的一种有用方法,可能有助于阐明疾病的分子机制或发现有用的疾病生物标志物。然而,在这种复杂组织中进行的分析的敏感性和可解释性受到其动态细胞异质性的显著影响。因此,需要对这种异质性进行量化,以便对其进行解释或更好地模拟某些转录本丰度、特定细胞类型与所研究指征之间可能存在的相互作用。然而,准确计数构成外周全血的多种组成细胞类型会使样本采集过程更加复杂,并导致额外成本。已经开发了许多方法来从高维转录组数据以及最近的表观遗传数据推断样本的组成。这些方法依赖于待计数细胞类型的分离表达谱的可用性。这些谱是平台特异性的,合适的数据集很少,并且生成它们成本很高。Affymetrix Gene ST平台上不存在这样的数据集。
我们展示了“Enumerateblood”,这是一个免费的开源R包,它公开了一个多响应高斯模型,能够根据Affymetrix Gene ST表达谱准确预测外周全血样本的组成,在应用于Gene ST数据时优于其他现有方法。
“Enumerateblood”显著提高了我们通过在流行的Affymetrix Gene ST平台上进行全血基因表达研究疾病病理生物学的能力,通过允许对这种复杂组织的各种成分进行更完整的研究而无需额外的数据收集。该模型的未来应用可能会从目前在基因表达综合数据库(GEO)网站上可用的约400个Affymetrix Gene ST血液基因表达数据集中产生新的见解。