Aviat Félix, Levitt Antoine, Stamm Benjamin, Maday Yvon, Ren Pengyu, Ponder Jay W, Lagardère Louis, Piquemal Jean-Philip
Laboratoire de Chimie Théorique, UPMC Université Paris 06, UMR 7617 , F-75005, Paris, France.
Inria Paris, F-75589 Paris Cedex 12, Université Paris-Est, CERMICS (ENPC) , Marne-la-Vallée, F-77455, France.
J Chem Theory Comput. 2017 Jan 10;13(1):180-190. doi: 10.1021/acs.jctc.6b00981. Epub 2016 Dec 8.
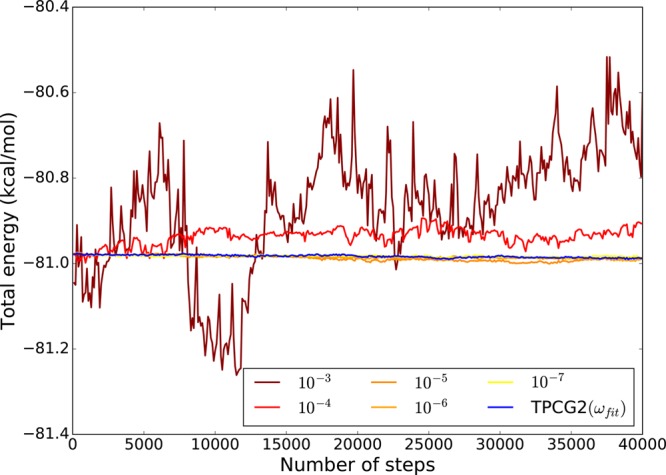
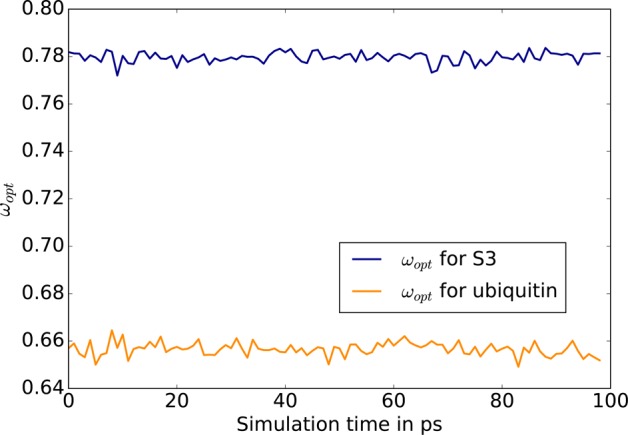
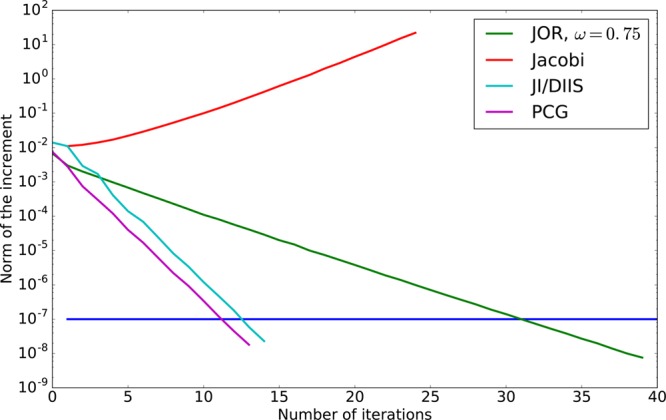
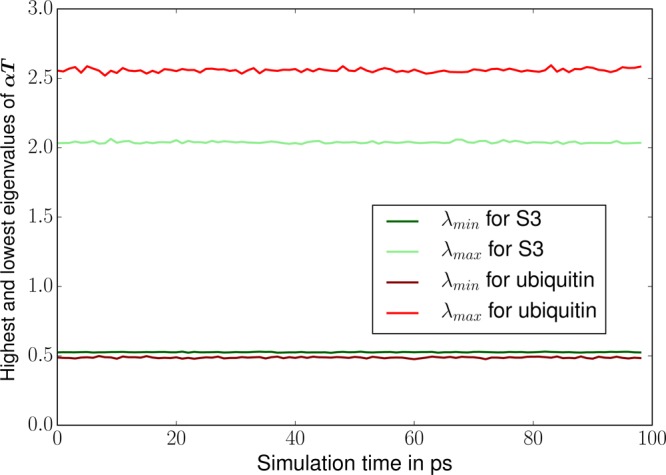
We introduce a new class of methods, denoted as Truncated Conjugate Gradient(TCG), to solve the many-body polarization energy and its associated forces in molecular simulations (i.e. molecular dynamics (MD) and Monte Carlo). The method consists in a fixed number of Conjugate Gradient (CG) iterations. TCG approaches provide a scalable solution to the polarization problem at a user-chosen cost and a corresponding optimal accuracy. The optimality of the CG-method guarantees that the number of the required matrix-vector products are reduced to a minimum compared to other iterative methods. This family of methods is non-empirical, fully adaptive, and provides analytical gradients, avoiding therefore any energy drift in MD as compared to popular iterative solvers. Besides speed, one great advantage of this class of approximate methods is that their accuracy is systematically improvable. Indeed, as the CG-method is a Krylov subspace method, the associated error is monotonically reduced at each iteration. On top of that, two improvements can be proposed at virtually no cost: (i) the use of preconditioners can be employed, which leads to the Truncated Preconditioned Conjugate Gradient (TPCG); (ii) since the residual of the final step of the CG-method is available, one additional Picard fixed point iteration ("peek"), equivalent to one step of Jacobi Over Relaxation (JOR) with relaxation parameter ω, can be made at almost no cost. This method is denoted by TCG-n(ω). Black-box adaptive methods to find good choices of ω are provided and discussed. Results show that TPCG-3(ω) is converged to high accuracy (a few kcal/mol) for various types of systems including proteins and highly charged systems at the fixed cost of four matrix-vector products: three CG iterations plus the initial CG descent direction. Alternatively, T(P)CG-2(ω) provides robust results at a reduced cost (three matrix-vector products) and offers new perspectives for long polarizable MD as a production algorithm. The T(P)CG-1(ω) level provides less accurate solutions for inhomogeneous systems, but its applicability to well-conditioned problems such as water is remarkable, with only two matrix-vector product evaluations.
我们引入了一类新的方法,称为截断共轭梯度法(TCG),用于在分子模拟(即分子动力学(MD)和蒙特卡罗模拟)中求解多体极化能及其相关力。该方法由固定次数的共轭梯度(CG)迭代组成。TCG方法以用户选择的成本和相应的最优精度为极化问题提供了一种可扩展的解决方案。CG方法的最优性保证了与其他迭代方法相比,所需的矩阵 - 向量乘积的数量减少到最小。这一类方法是非经验性的、完全自适应的,并提供解析梯度,因此与流行的迭代求解器相比,避免了MD中的任何能量漂移。除了速度之外,这类近似方法的一个很大优点是它们的精度可以系统地提高。实际上,由于CG方法是一种Krylov子空间方法,每次迭代时相关误差都会单调减小。除此之外,几乎无需成本就可以提出两种改进方法:(i)可以使用预处理器,这就产生了截断预处理共轭梯度法(TPCG);(ii)由于CG方法最后一步的残差是可用的,几乎无需成本就可以进行一次额外的皮卡德不动点迭代(“窥探”),这相当于具有松弛参数ω的雅可比超松弛(JOR)的一步。这种方法表示为TCG - n(ω)。我们提供并讨论了用于找到ω的良好选择的黑箱自适应方法。结果表明,对于包括蛋白质和高电荷系统在内的各种类型的系统,TPCG - 3(ω)以四个矩阵 - 向量乘积的固定成本收敛到高精度(几千卡/摩尔):三次CG迭代加上初始CG下降方向。或者,T(P)CG - 2(ω)以降低的成本(三个矩阵 - 向量乘积)提供了稳健的结果,并为作为一种生产算法的长时间极化MD提供了新的视角。T(P)CG - 1(ω)水平为非均匀系统提供的解精度较低,但其在诸如水等条件良好的问题上的适用性非常显著,只需要两次矩阵 - 向量乘积评估。