Hatch Robert John, Wei Yan, Xia Di, Götz Jürgen
Clem Jones Centre for Ageing Dementia Research, Queensland Brain Institute, The University of Queensland, St Lucia Campus, Brisbane, QLD, 4072, Australia.
State Key Laboratory of Brain and Cognitive Science, Institute of Biophysics, Chinese Academy of Sciences, Beijing, 100101, China.
Acta Neuropathol. 2017 May;133(5):717-730. doi: 10.1007/s00401-017-1674-1. Epub 2017 Jan 16.
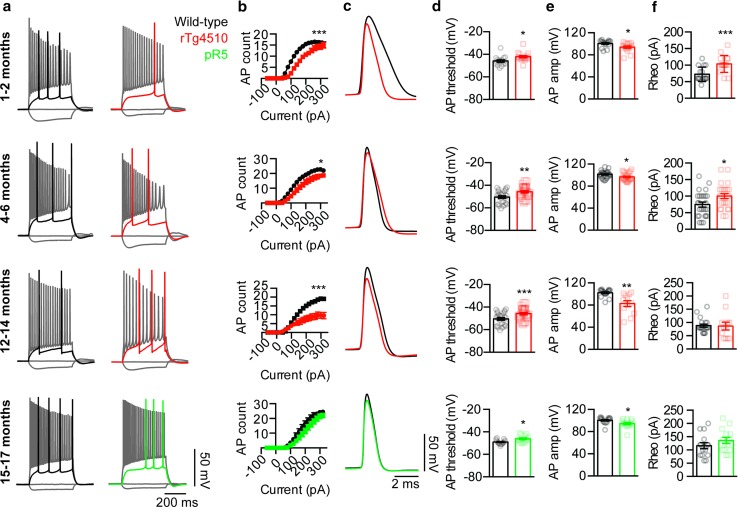
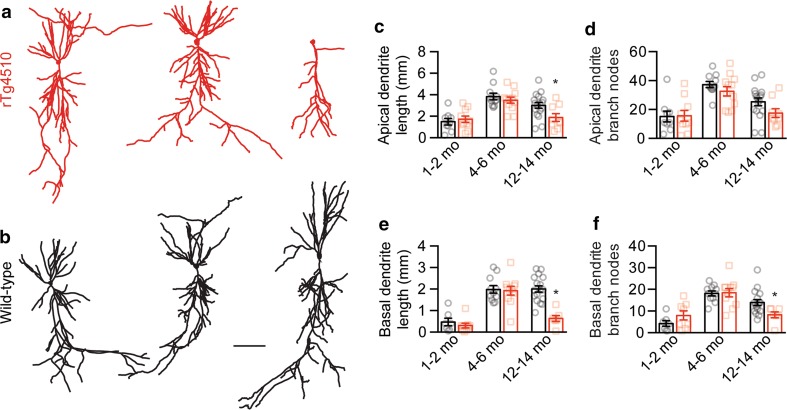
Hyperphosphorylated tau has a critical role in tauopathies such as Alzheimer's disease and frontotemporal dementia, impairing neuronal function and eventually leading to neurodegeneration. A critical role for tau is supported by studies in transgenic mouse models that express the P301L tau mutation found in cases of familial frontotemporal dementia, with the accumulation of hyperphosphorylated tau in the hippocampus causing reductions in hippocampal long-term potentiation and impairments in spatial learning and memory. However, what has remained unexplored is the role of hyperphosphorylated tau in reducing neuronal excitability. Here, we show in two complementary P301L tau transgenic mouse models that hyperphosphorylated tau induces a more depolarized threshold for action potential initiation and reduces firing in hippocampal CA1 neurons, which was rescued by the suppression of transgenic tau. Furthermore, using mutagenesis and primary hippocampal neuronal cultures, we reveal that this reduction in neuronal excitability results from the relocation of the axon initial segment (AIS) down the axon in a tau phosphorylation-dependent manner. We also demonstrate that this effect is microtubule-dependent. In addition, pharmacological stabilization was found to prevent both the structural and functional deficits caused by tau hyperphosphorylation. Finally, we demonstrate that the AIS of neurons from tau transgenic mice is further down the axon, which correlates with a reduction in excitability. We therefore propose that a reduction in hippocampal excitability due to a tau-mediated distal relocalization of the AIS contributes to the hippocampal dysfunction observed in tauopathies.
过度磷酸化的tau蛋白在诸如阿尔茨海默病和额颞叶痴呆等tau蛋白病中起着关键作用,它会损害神经元功能并最终导致神经退行性变。在转基因小鼠模型中的研究支持了tau蛋白的关键作用,这些模型表达了在家族性额颞叶痴呆病例中发现的P301L tau突变,海马体中过度磷酸化tau蛋白的积累导致海马体长期增强作用降低以及空间学习和记忆受损。然而,过度磷酸化的tau蛋白在降低神经元兴奋性方面的作用尚未得到探索。在此,我们在两种互补的P301L tau转基因小鼠模型中表明,过度磷酸化的tau蛋白会诱导动作电位起始的阈值更去极化,并降低海马CA1神经元的放电频率,而抑制转基因tau蛋白可使其恢复。此外,通过诱变和原代海马神经元培养,我们揭示这种神经元兴奋性的降低是由于轴突起始段(AIS)以tau蛋白磷酸化依赖的方式向轴突下游重新定位所致。我们还证明这种作用是微管依赖的。另外,发现药物稳定作用可预防由tau蛋白过度磷酸化引起的结构和功能缺陷。最后,我们证明来自tau转基因小鼠的神经元的AIS在轴突上更靠下游,这与兴奋性降低相关。因此,我们提出由于tau介导的AIS向远端重新定位导致的海马兴奋性降低促成了在tau蛋白病中观察到的海马功能障碍。