Siriphap Achiraya, Leekitcharoenphon Pimlapas, Kaas Rolf S, Theethakaew Chonchanok, Aarestrup Frank M, Sutheinkul Orasa, Hendriksen Rene S
Department of Microbiology, Faculty of Public Health, Mahidol University, Bangkok, Thailand.
National Food Institute, Technical University of Denmark, Research Group for Genomic Epidemiology, WHO Collaborating Center for Antimicrobial Resistance in Foodborne Pathogens and Genomics and European Union Reference Laboratory for Antimicrobial Resistance, Kgs. Lyngby, Denmark.
PLoS One. 2017 Jan 19;12(1):e0169324. doi: 10.1371/journal.pone.0169324. eCollection 2017.
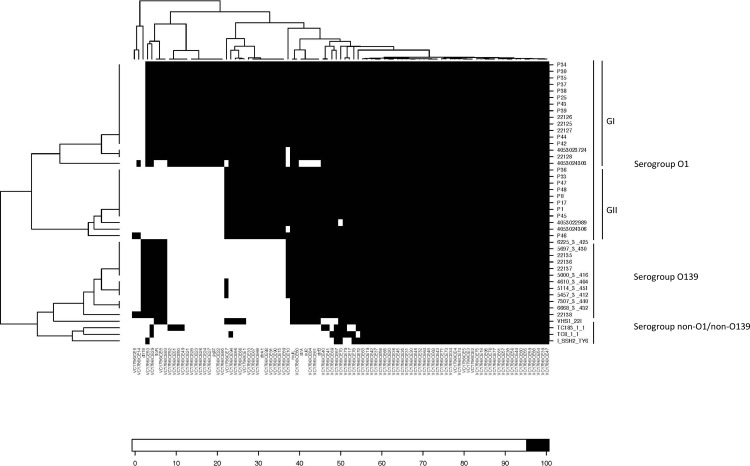
Cholera is still an important public health problem in several countries, including Thailand. In this study, a collection of clinical and environmental V. cholerae serogroup O1, O139, and non-O1/non-O139 strains originating from Thailand (1983 to 2013) was characterized to determine phenotypic and genotypic traits and to investigate the genetic relatedness. Using a combination of conventional methods and whole genome sequencing (WGS), 78 V. cholerae strains were identified. WGS was used to determine the serogroup, biotype, virulence, mobile genetic elements, and antimicrobial resistance genes using online bioinformatics tools. In addition, phenotypic antimicrobial resistance was determined by the minimal inhibitory concentration (MIC) test. The 78 V. cholerae strains belonged to the following serogroups O1: (n = 44), O139 (n = 16) and non-O1/non-O139 (n = 18). Interestingly, we found that the typical El Tor O1 strains were the major cause of clinical cholera during 1983-2000 with two Classical O1 strains detected in 2000. In 2004-2010, the El Tor variant strains revealed genotypes of the Classical biotype possessing either only ctxB or both ctxB and rstR while they harbored tcpA of the El Tor biotype. Thirty O1 and eleven O139 clinical strains carried CTXϕ (Cholera toxin) and tcpA as well four different pathogenic islands (PAIs). Beside non-O1/non-O139, the O1 environmental strains also presented chxA and Type Three Secretion System (TTSS). The in silico MultiLocus Sequence Typing (MLST) discriminated the O1 and O139 clinical strains from other serogroups and environmental strains. ST69 was dominant in the clinical strains belonging to the 7th pandemic clone. Non-O1/non-O139 and environmental strains showed various novel STs indicating genetic variation. Multidrug-resistant (MDR) strains were observed and conferred resistance to ampicillin, azithromycin, nalidixic acid, sulfamethoxazole, tetracycline, and trimethoprim and harboured variants of the SXT elements. For the first time since 1986, the presence of V. cholerae O1 Classical was reported causing cholera outbreaks in Thailand. In addition, we found that V. cholerae O1 El Tor variant and O139 were pre-dominating the pathogenic strains in Thailand. Using WGS and bioinformatic tools to analyze both historical and contemporary V. cholerae circulating in Thailand provided a more detailed understanding of the V. cholerae epidemiology, which ultimately could be applied for control measures and management of cholera in Thailand.
霍乱在包括泰国在内的几个国家仍然是一个重要的公共卫生问题。在本研究中,对1983年至2013年源自泰国的霍乱弧菌O1群、O139群以及非O1/非O139群的临床和环境菌株进行了特征分析,以确定其表型和基因型特征,并研究其遗传相关性。采用传统方法与全基因组测序(WGS)相结合的方式,鉴定出78株霍乱弧菌菌株。利用在线生物信息学工具,通过WGS确定血清群、生物型、毒力、可移动遗传元件和抗菌耐药基因。此外,通过最低抑菌浓度(MIC)试验确定表型抗菌耐药性。这78株霍乱弧菌菌株属于以下血清群:O1群(n = 44)、O139群(n = 16)和非O1/非O139群(n = 18)。有趣的是,我们发现典型的埃尔托O1菌株是1983 - 2000年期间临床霍乱的主要病因,2000年检测到两株古典O1菌株。在2004 - 2010年期间,埃尔托变异菌株显示出仅携带ctxB或同时携带ctxB和rstR的古典生物型基因型,而它们携带的是埃尔托生物型的tcpA。30株O1群和11株O139群临床菌株携带霍乱毒素(CTXϕ)和tcpA以及四种不同的致病岛(PAIs)。除了非O1/非O139群菌株外,O1群环境菌株也呈现出chxA和三型分泌系统(TTSS)。通过电子多基因座序列分型(MLST)将O1群和O139群临床菌株与其他血清群及环境菌株区分开来。ST69在属于第七次大流行克隆的临床菌株中占主导地位。非O1/非O139群和环境菌株显示出各种新的序列型(STs),表明存在遗传变异。观察到多药耐药(MDR)菌株,它们对氨苄西林、阿奇霉素、萘啶酸、磺胺甲恶唑、四环素和甲氧苄啶具有耐药性,并携带SXT元件的变体。自1986年以来首次报道了霍乱弧菌O1古典生物型在泰国引发霍乱疫情。此外,我们发现霍乱弧菌O1埃尔托变异株和O139群在泰国的致病菌株中占主导地位。利用WGS和生物信息学工具分析泰国历史上和当代流行的霍乱弧菌,能更详细地了解霍乱弧菌的流行病学情况,这最终可应用于泰国霍乱的控制措施和管理。