Department of Biostatistics and Computational Biology.
Department of Microbiology and Immunology, University of Rochester, Rochester, NY 14642, USA.
Bioinformatics. 2017 Jul 1;33(13):1944-1952. doi: 10.1093/bioinformatics/btx104.
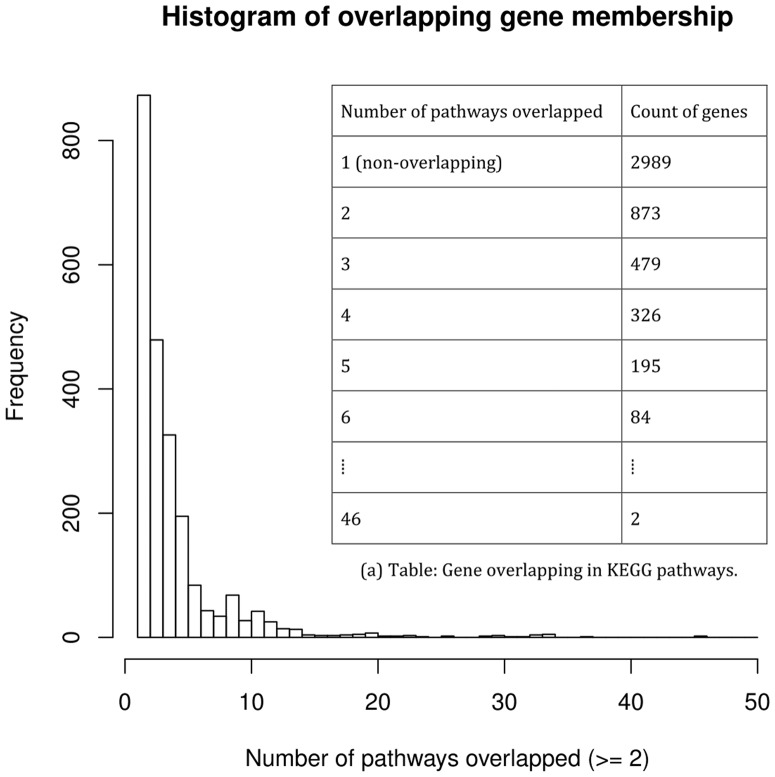
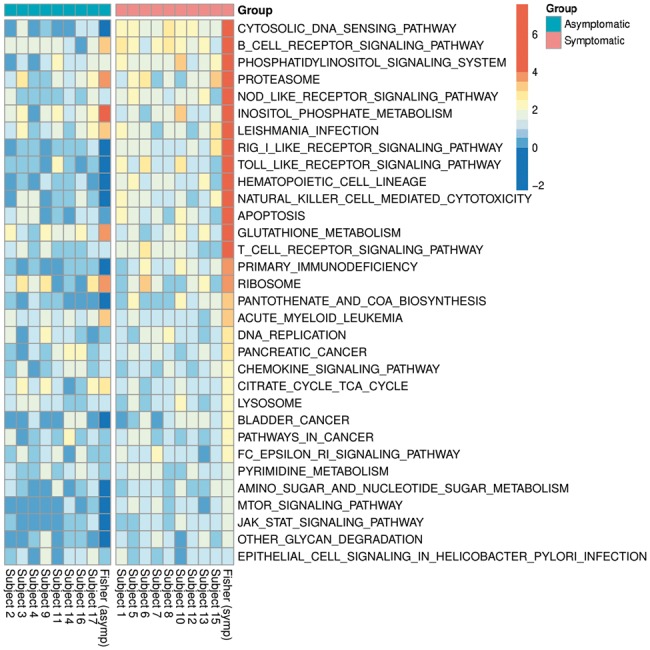
Gene set enrichment analyses (GSEAs) are widely used in genomic research to identify underlying biological mechanisms (defined by the gene sets), such as Gene Ontology terms and molecular pathways. There are two caveats in the currently available methods: (i) they are typically designed for group comparisons or regression analyses, which do not utilize temporal information efficiently in time-series of transcriptomics measurements; and (ii) genes overlapping in multiple molecular pathways are considered multiple times in hypothesis testing.
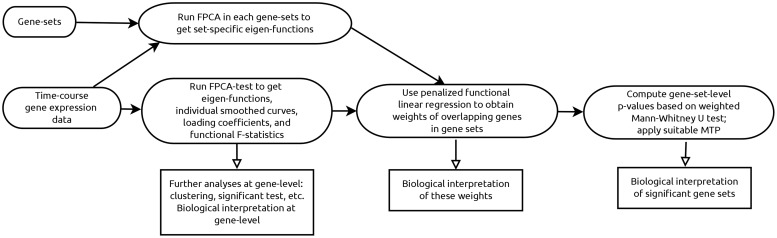
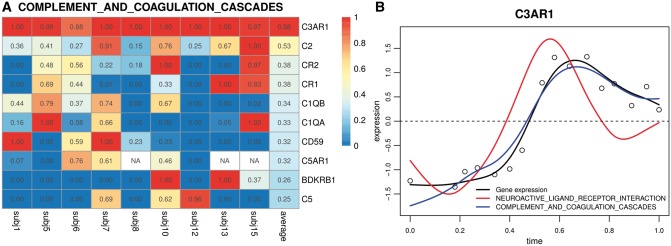
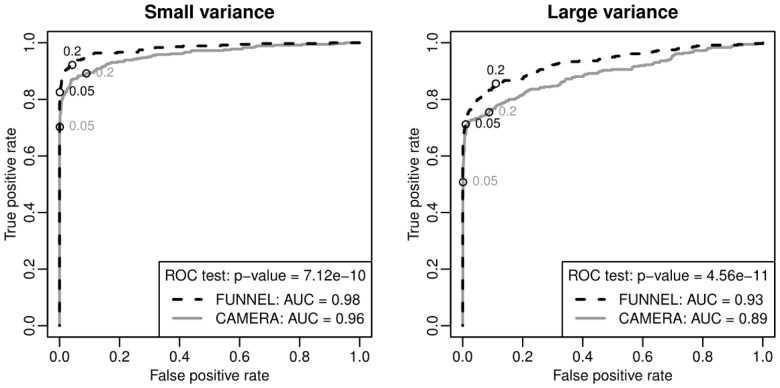
We propose an inferential framework for GSEA based on functional data analysis, which utilizes the temporal information based on functional principal component analysis, and disentangles the effects of overlapping genes by a functional extension of the elastic-net regression. Furthermore, the hypothesis testing for the gene sets is performed by an extension of Mann-Whitney U test which is based on weighted rank sums computed from correlated observations. By using both simulated datasets and a large-scale time-course gene expression data on human influenza infection, we demonstrate that our method has uniformly better receiver operating characteristic curves, and identifies more pathways relevant to immune-response to human influenza infection than the competing approaches.
The methods are implemented in R package FUNNEL, freely and publicly available at: https://github.com/yunzhang813/FUNNEL-GSEA-R-Package .
xing_qiu@urmc.rochester.edu or juilee_thakar@urmc.rochester.edu.
Supplementary data are available at Bioinformatics online.
基因集富集分析(GSEA)广泛应用于基因组研究中,以识别潜在的生物学机制(由基因集定义),如基因本体论术语和分子途径。目前可用方法存在两个问题:(i)它们通常是为组比较或回归分析设计的,在转录组测量的时间序列中不能有效地利用时间信息;(ii)在多个分子途径中重叠的基因在假设检验中被多次考虑。
我们提出了一种基于功能数据分析的 GSEA 推断框架,该框架利用基于功能主成分分析的时间信息,并通过弹性网络回归的功能扩展来分解重叠基因的影响。此外,通过基于相关观测的加权秩和计算的 Mann-Whitney U 检验的扩展,对基因集进行假设检验。通过使用模拟数据集和大规模的人类流感感染时间过程基因表达数据,我们证明我们的方法具有均匀更好的接收器操作特征曲线,并且比竞争方法识别出更多与人类流感感染免疫反应相关的途径。
该方法在 R 包 FUNNEL 中实现,可在以下网址免费公开获得:https://github.com/yunzhang813/FUNNEL-GSEA-R-Package。
xing_qiu@urmc.rochester.edu 或 juilee_thakar@urmc.rochester.edu。
补充数据可在生物信息学在线获得。