MRC-University of Glasgow Centre For Virus Research, Glasgow, United Kingdom.
School of Computing Science, University of Glasgow, Glasgow, United Kingdom.
PLoS Comput Biol. 2020 May 26;16(5):e1007894. doi: 10.1371/journal.pcbi.1007894. eCollection 2020 May.
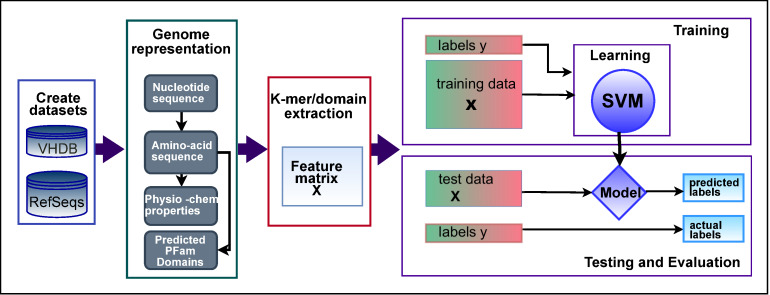
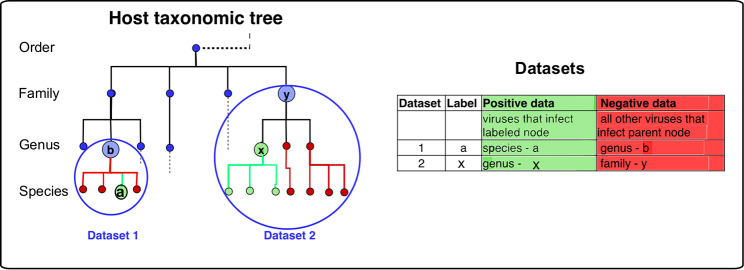
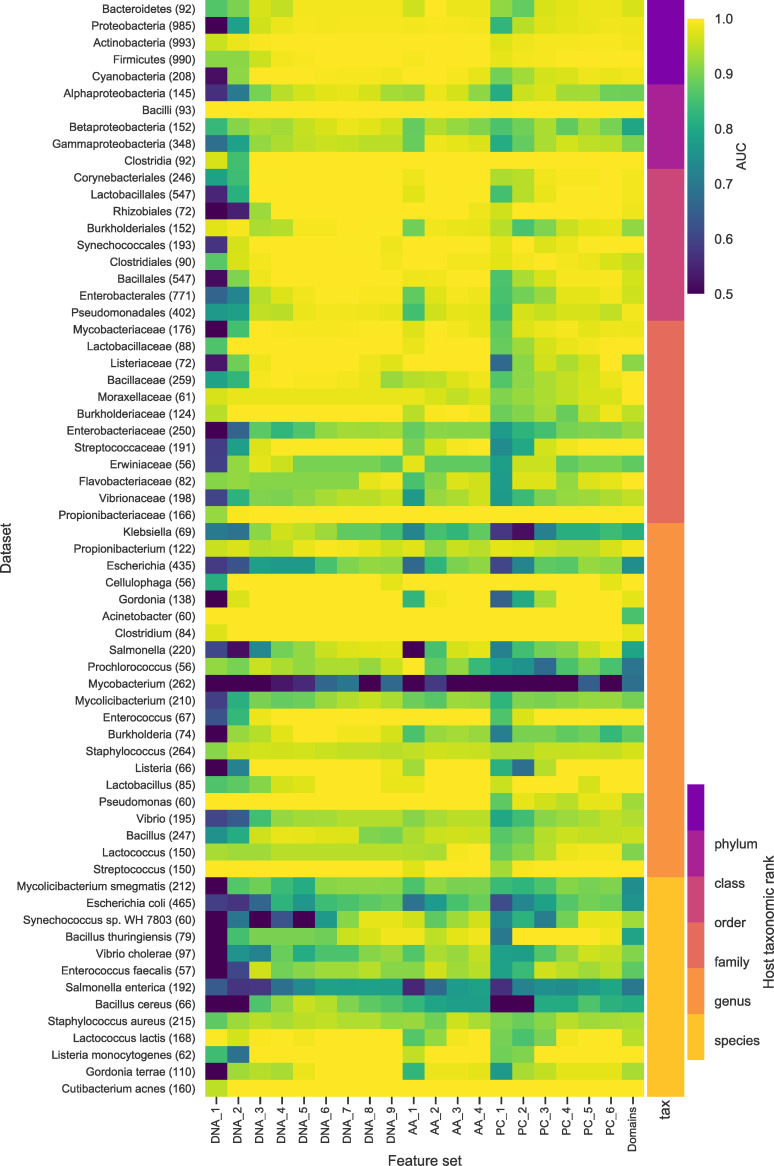
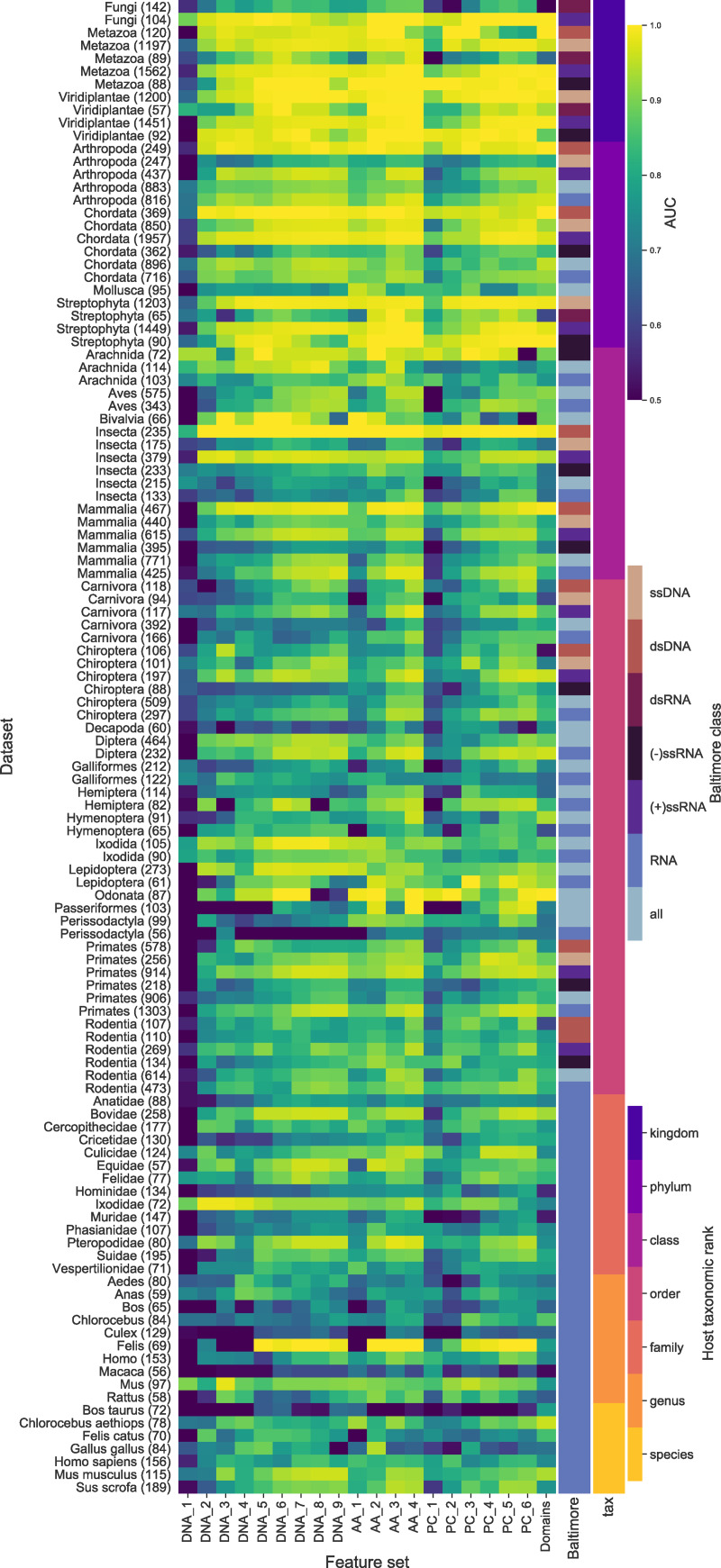
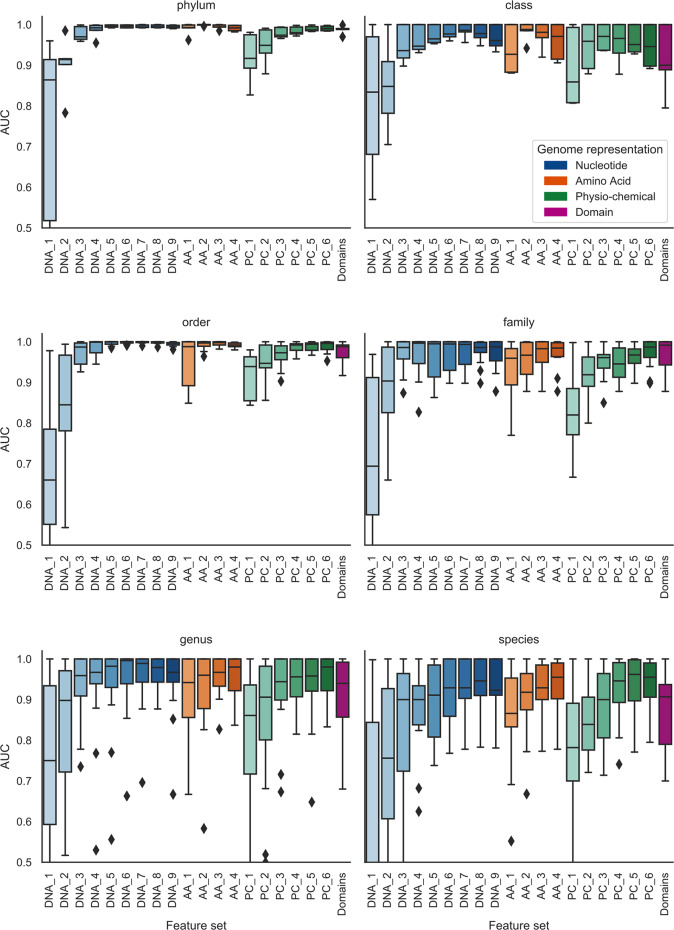
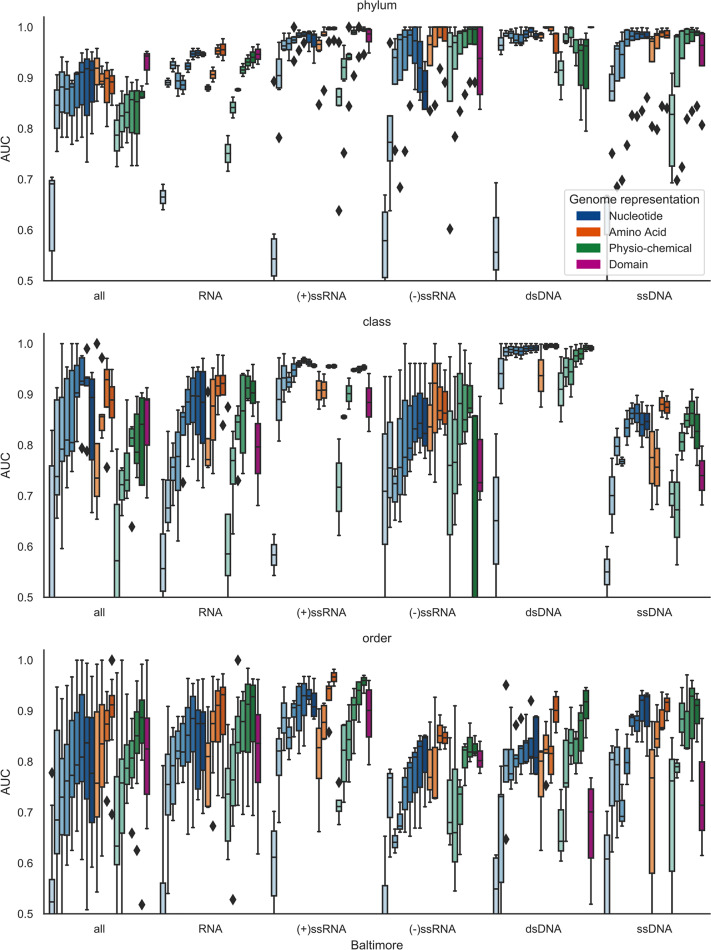
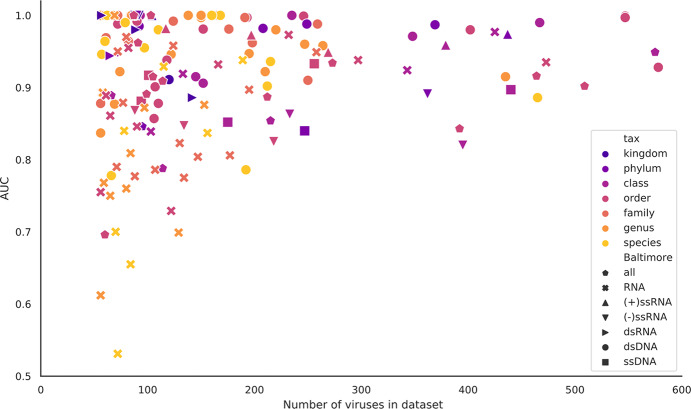
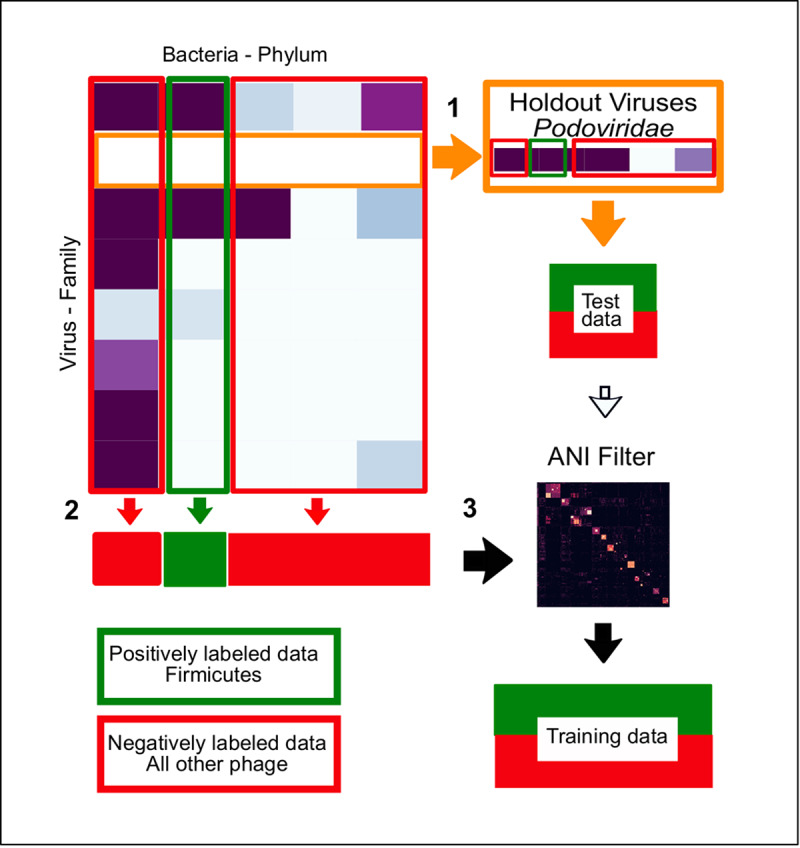
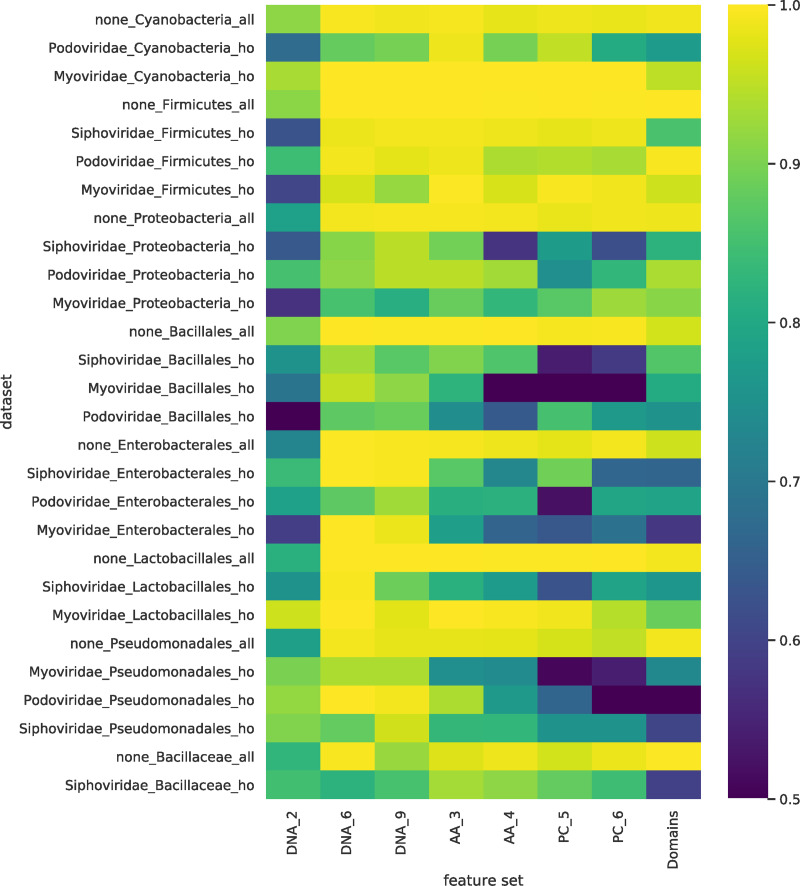
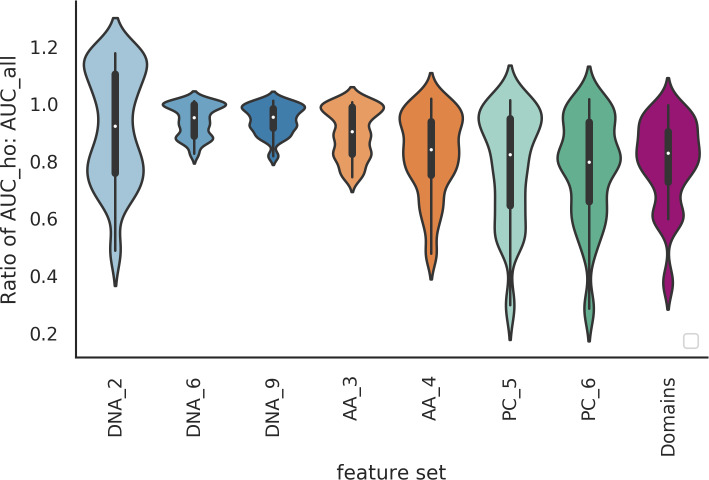
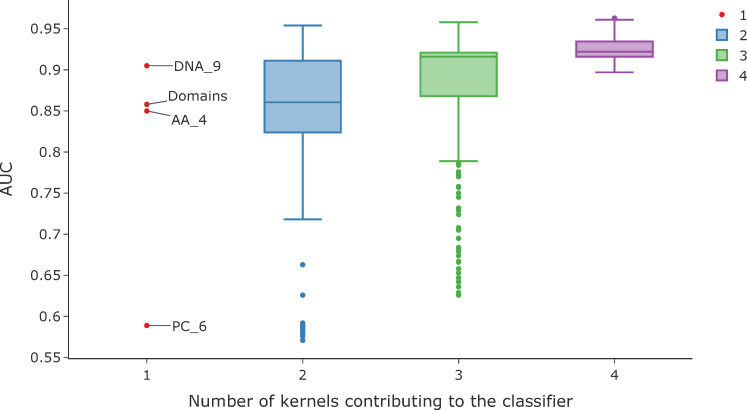
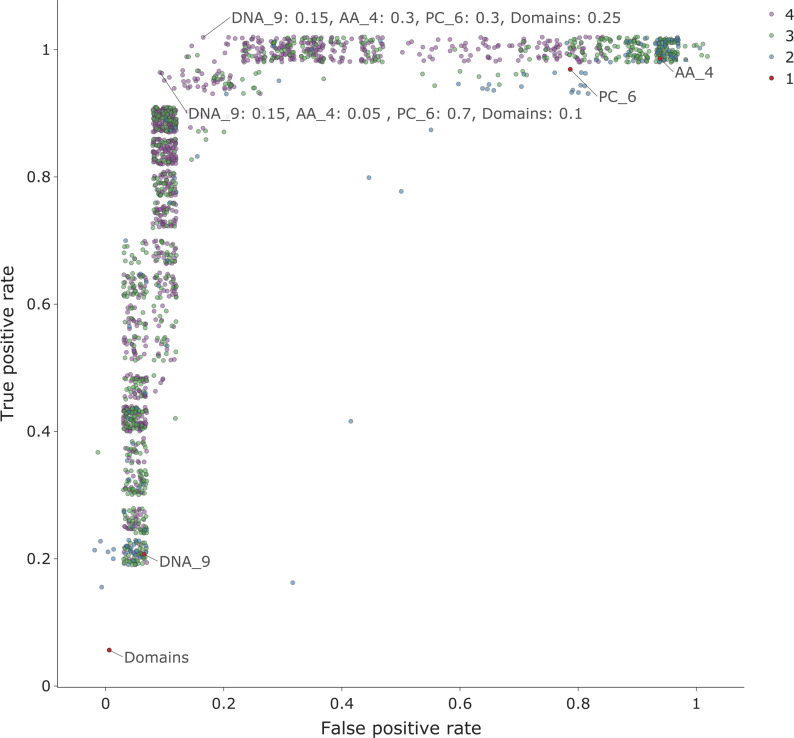
The rise in metagenomics has led to an exponential growth in virus discovery. However, the majority of these new virus sequences have no assigned host. Current machine learning approaches to predicting virus host interactions have a tendency to focus on nucleotide features, ignoring other representations of genomic information. Here we investigate the predictive potential of features generated from four different 'levels' of viral genome representation: nucleotide, amino acid, amino acid properties and protein domains. This more fully exploits the biological information present in the virus genomes. Over a hundred and eighty binary datasets for infecting versus non-infecting viruses at all taxonomic ranks of both eukaryote and prokaryote hosts were compiled. The viral genomes were converted into the four different levels of genome representation and twenty feature sets were generated by extracting k-mer compositions and predicted protein domains. We trained and tested Support Vector Machine, SVM, classifiers to compare the predictive capacity of each of these feature sets for each dataset. Our results show that all levels of genome representation are consistently predictive of host taxonomy and that prediction k-mer composition improves with increasing k-mer length for all k-mer based features. Using a phylogenetically aware holdout method, we demonstrate that the predictive feature sets contain signals reflecting both the evolutionary relationship between the viruses infecting related hosts, and host-mimicry. Our results demonstrate that incorporating a range of complementary features, generated purely from virus genome sequences, leads to improved accuracy for a range of virus host prediction tasks enabling computational assignment of host taxonomic information.
宏基因组学的兴起导致了病毒发现的指数级增长。然而,这些新的病毒序列大多数都没有指定的宿主。目前用于预测病毒宿主相互作用的机器学习方法往往侧重于核苷酸特征,而忽略了基因组信息的其他表示形式。在这里,我们研究了从病毒基因组表示的四个不同“层次”生成的特征的预测潜力:核苷酸、氨基酸、氨基酸性质和蛋白质结构域。这更充分地利用了病毒基因组中存在的生物学信息。我们收集了一百八十多个二元数据集,涵盖了真核生物和原核生物宿主的所有分类等级的感染性和非感染性病毒,这些数据集用于比较预测病毒宿主相互作用的特征集。我们将病毒基因组转换为四个不同的基因组表示层次,并通过提取 k-mer 组成和预测的蛋白质结构域生成了二十个特征集。我们训练和测试了支持向量机 (SVM) 分类器,以比较这些特征集中的每一个对每个数据集的预测能力。我们的结果表明,所有的基因组表示层次都能一致地预测宿主的分类学,并且随着 k-mer 长度的增加,基于 k-mer 的特征的预测性能也会提高。使用一种基于系统发育的保留方法,我们证明了预测特征集包含反映感染相关宿主的病毒之间进化关系的信号,以及宿主模拟。我们的结果表明,将一系列互补的特征(纯粹从病毒基因组序列中生成)结合起来,可以提高一系列病毒宿主预测任务的准确性,从而实现宿主分类信息的计算分配。