Center for Bioinformatics and Systems Biology and Department of Radiology, Wake Forest School of Medicine, Winston-Salem, NC, 27157, USA.
Sci Rep. 2017 Apr 4;7(1):614. doi: 10.1038/s41598-017-00795-4.
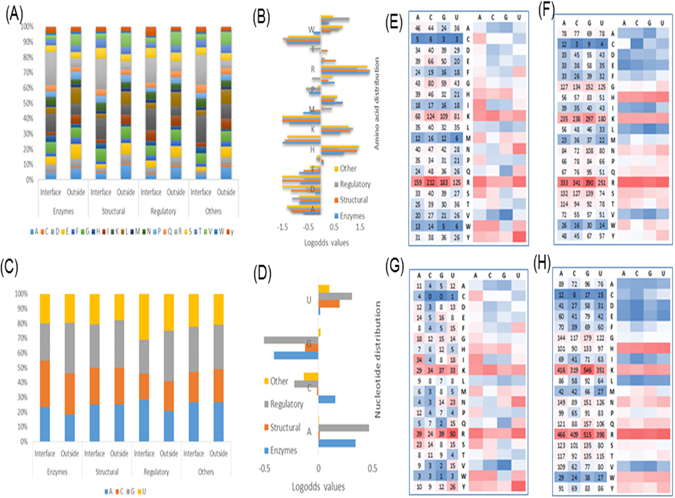
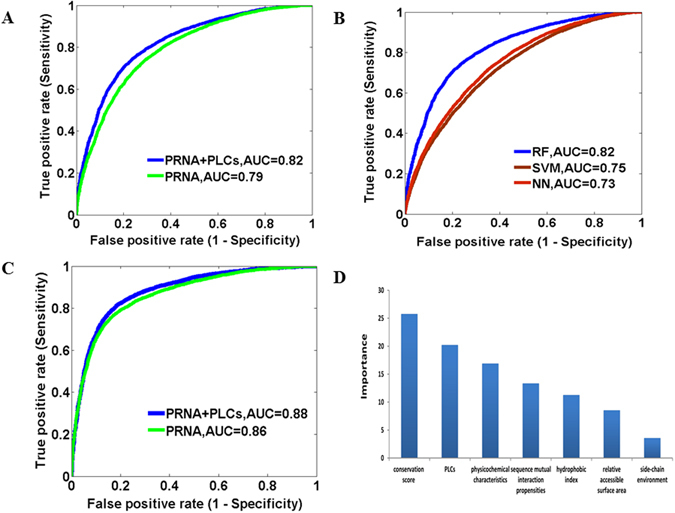
RNA and protein interactions play crucial roles in multiple biological processes, while these interactions are significantly influenced by the structures and sequences of protein and RNA molecules. In this study, we first performed an analysis of RNA-protein interacting complexes, and identified interface properties of sequences and structures, which reveal the diverse nature of the binding sites. With the observations, we built a three-step prediction model, namely RPI-Bind, for the identification of RNA-protein binding regions using the sequences and structures of both proteins and RNAs. The three steps include 1) the prediction of RNA binding regions on protein, 2) the prediction of protein binding regions on RNA, and 3) the prediction of interacting regions on both RNA and protein simultaneously, with the results from steps 1) and 2). Compared with existing methods, most of which employ only sequences, our model significantly improves the prediction accuracy at each of the three steps. Especially, our model outperforms the catRAPID by >20% at the 3 step. All of these results indicate the importance of structures in RNA-protein interactions, and suggest that the RPI-Bind model is a powerful theoretical framework for studying RNA-protein interactions.
RNA 和蛋白质相互作用在多种生物过程中起着至关重要的作用,而这些相互作用受到蛋白质和 RNA 分子的结构和序列的显著影响。在这项研究中,我们首先对 RNA-蛋白质相互作用复合物进行了分析,并确定了序列和结构的界面特性,这些特性揭示了结合位点的多样性。基于这些观察结果,我们构建了一个三步预测模型,即 RPI-Bind,用于使用蛋白质和 RNA 的序列和结构识别 RNA-蛋白质结合区域。这三个步骤包括 1)预测蛋白质上的 RNA 结合区域,2)预测 RNA 上的蛋白质结合区域,以及 3)同时预测 RNA 和蛋白质上的相互作用区域,结果来自步骤 1)和 2)。与现有的方法相比,这些方法大多只使用序列,我们的模型在这三个步骤中的每个步骤都显著提高了预测准确性。特别是,我们的模型在第三步中的表现优于 catRAPID 超过 20%。所有这些结果都表明结构在 RNA-蛋白质相互作用中的重要性,并表明 RPI-Bind 模型是研究 RNA-蛋白质相互作用的强大理论框架。