Smith R A, Ionides E L, King A A
Department of Bioinformatics, University of Michigan, Ann Arbor, MI.
Department of Statistics, University of Michigan, Ann Arbor, MI.
Mol Biol Evol. 2017 Aug 1;34(8):2065-2084. doi: 10.1093/molbev/msx124.
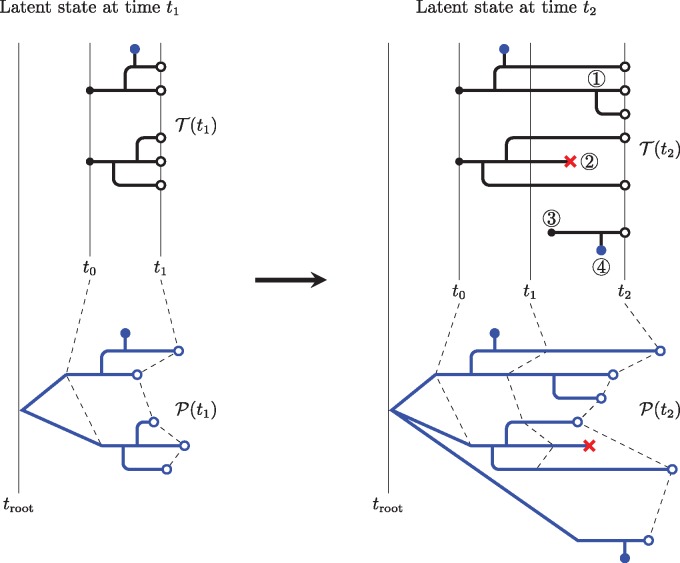
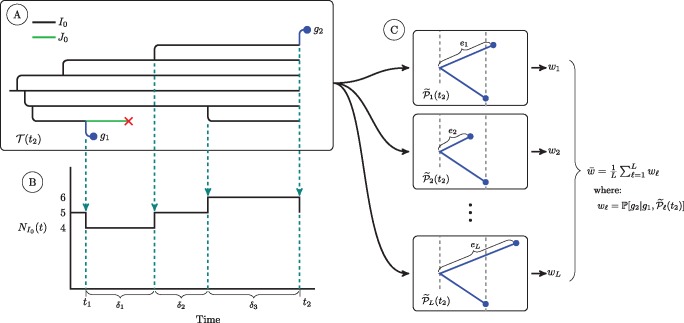
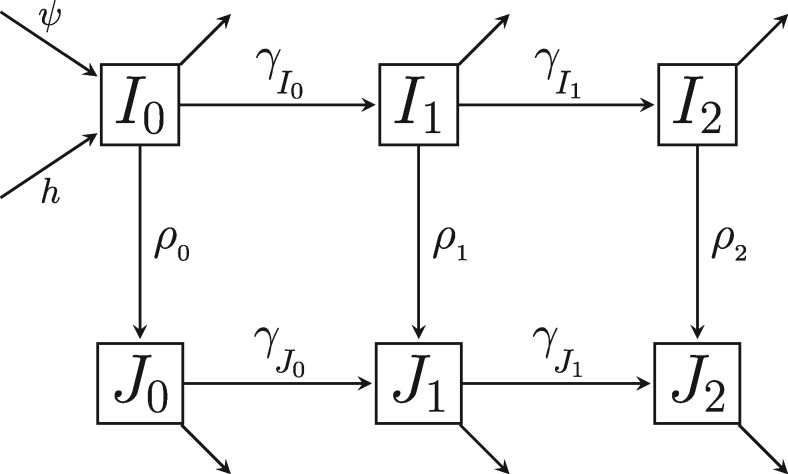
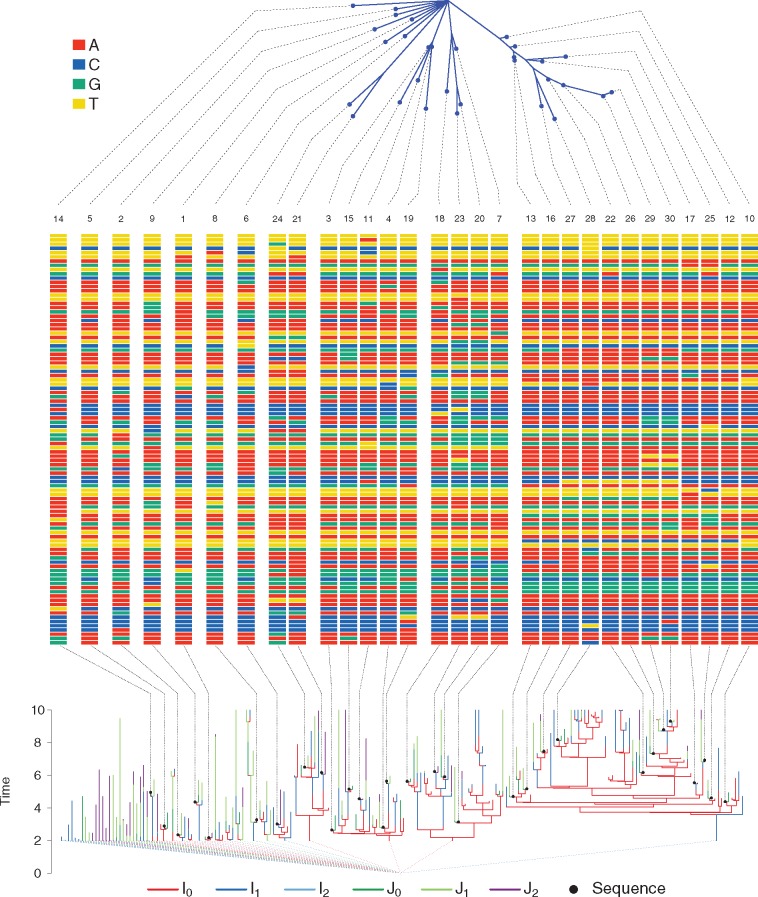
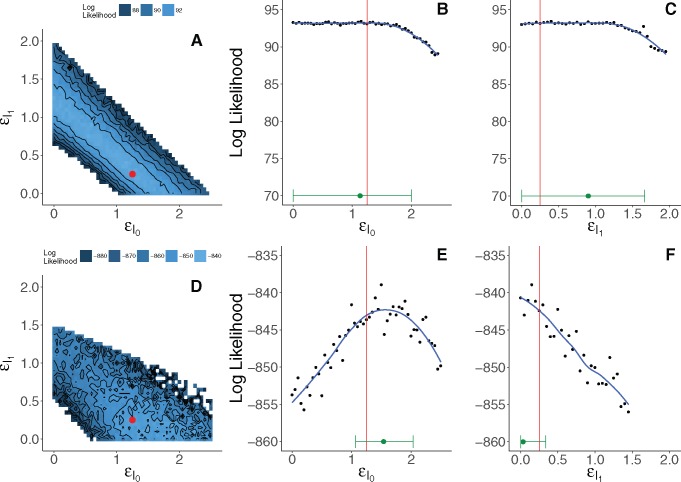
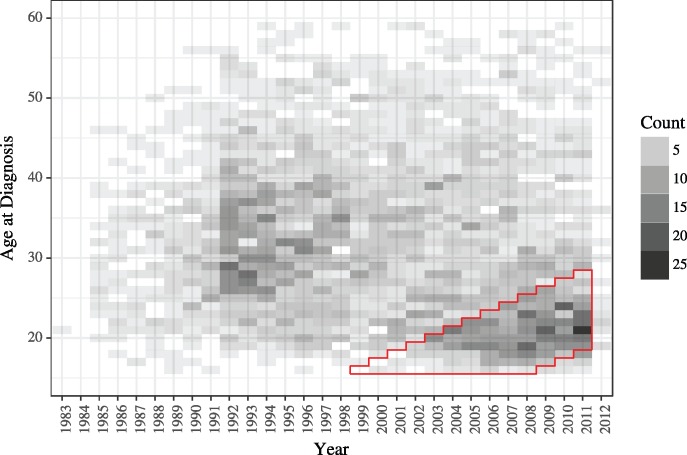
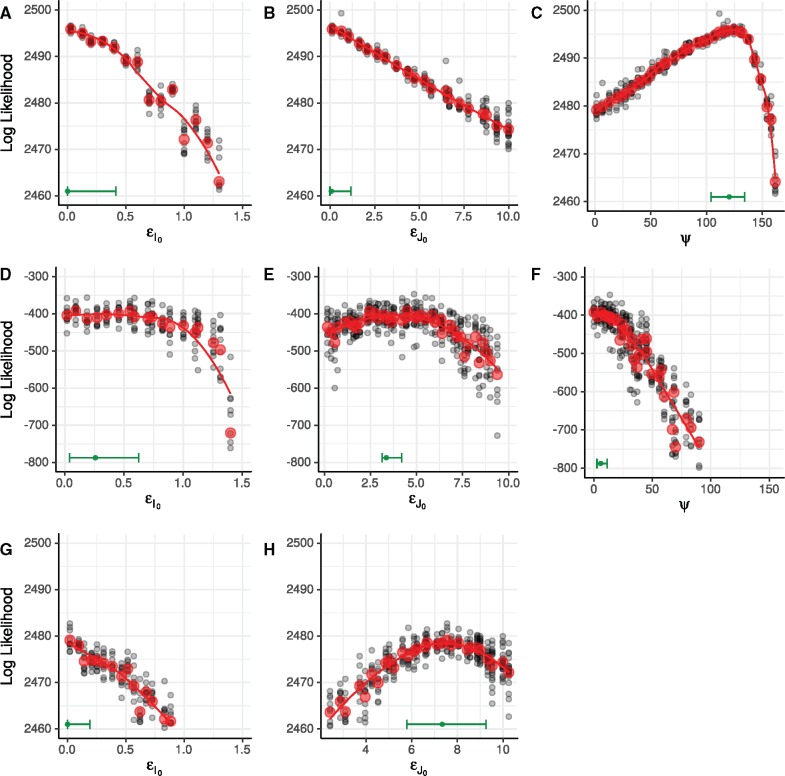
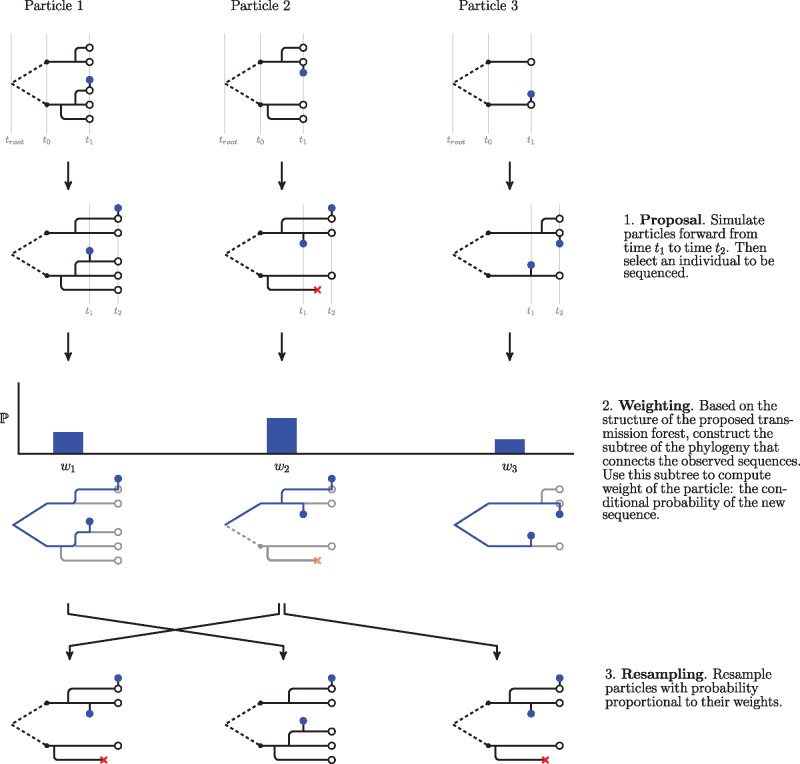
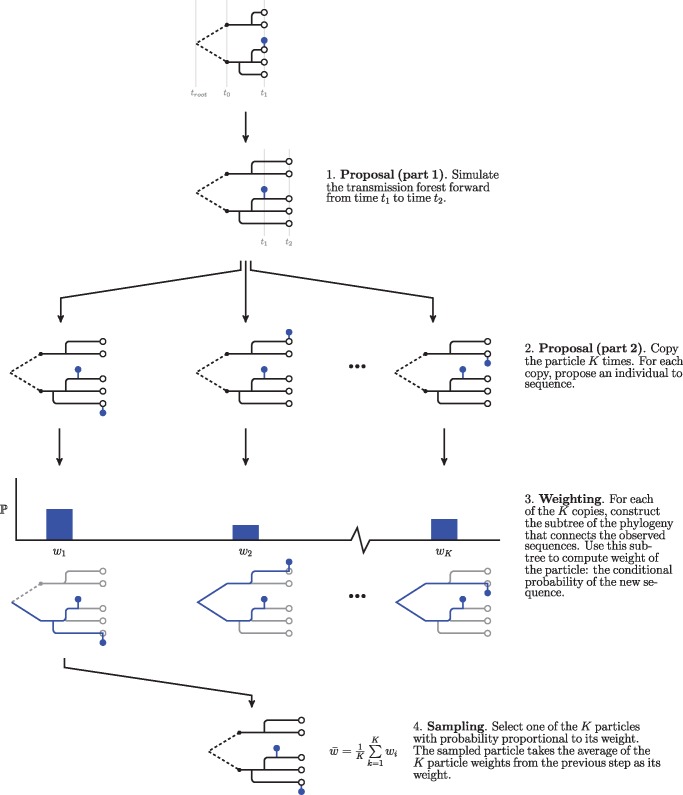
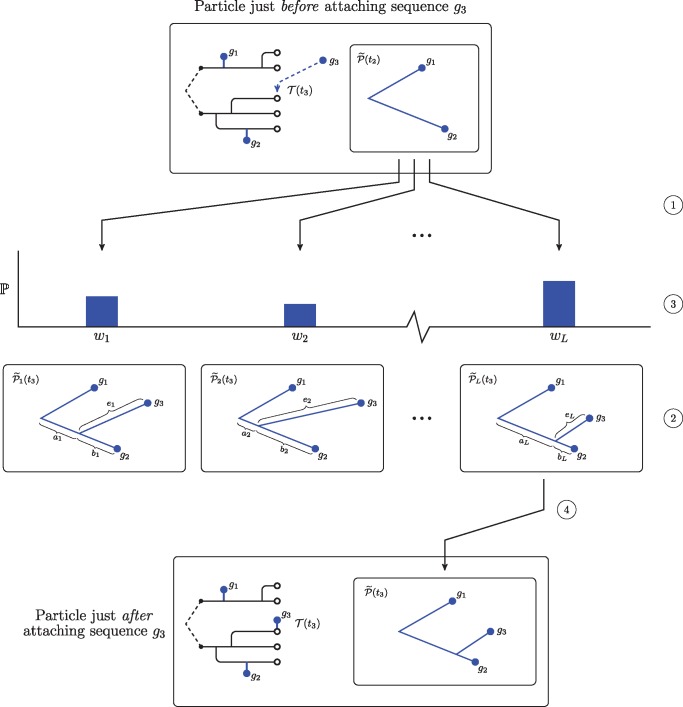
Genetic sequences from pathogens can provide information about infectious disease dynamics that may supplement or replace information from other epidemiological observations. Most currently available methods first estimate phylogenetic trees from sequence data, then estimate a transmission model conditional on these phylogenies. Outside limited classes of models, existing methods are unable to enforce logical consistency between the model of transmission and that underlying the phylogenetic reconstruction. Such conflicts in assumptions can lead to bias in the resulting inferences. Here, we develop a general, statistically efficient, plug-and-play method to jointly estimate both disease transmission and phylogeny using genetic data and, if desired, other epidemiological observations. This method explicitly connects the model of transmission and the model of phylogeny so as to avoid the aforementioned inconsistency. We demonstrate the feasibility of our approach through simulation and apply it to estimate stage-specific infectiousness in a subepidemic of human immunodeficiency virus in Detroit, Michigan. In a supplement, we prove that our approach is a valid sequential Monte Carlo algorithm. While we focus on how these methods may be applied to population-level models of infectious disease, their scope is more general. These methods may be applied in other biological systems where one seeks to infer population dynamics from genetic sequences, and they may also find application for evolutionary models with phenotypic rather than genotypic data.
病原体的基因序列能够提供有关传染病动态的信息,这些信息可能补充或取代来自其他流行病学观察的信息。目前大多数可用方法首先根据序列数据估计系统发育树,然后在这些系统发育的基础上估计传播模型。在有限的模型类别之外,现有方法无法在传播模型与系统发育重建所依据的模型之间强制实现逻辑一致性。这种假设上的冲突可能导致所得推断出现偏差。在此,我们开发了一种通用的、统计高效的即插即用方法,利用基因数据以及(如有需要)其他流行病学观察结果,共同估计疾病传播和系统发育。该方法明确地将传播模型与系统发育模型联系起来,以避免上述不一致性。我们通过模拟证明了我们方法的可行性,并将其应用于估计密歇根州底特律市人类免疫缺陷病毒局部流行中的特定阶段传染性。在附录中,我们证明了我们的方法是一种有效的序贯蒙特卡罗算法。虽然我们专注于这些方法如何应用于传染病的群体水平模型,但其适用范围更为广泛。这些方法可应用于其他生物系统,在这些系统中人们试图从基因序列推断群体动态,并且它们也可能适用于具有表型而非基因型数据的进化模型。