Cody Alison J, Bray James E, Jolley Keith A, McCarthy Noel D, Maiden Martin C J
Department of Zoology, University of Oxford, Oxford, United Kingdom
NIHR Health Protection Research Unit in Gastrointestinal Infections, University of Oxford, Oxford, United Kingdom.
J Clin Microbiol. 2017 Jul;55(7):2086-2097. doi: 10.1128/JCM.00080-17. Epub 2017 Apr 26.
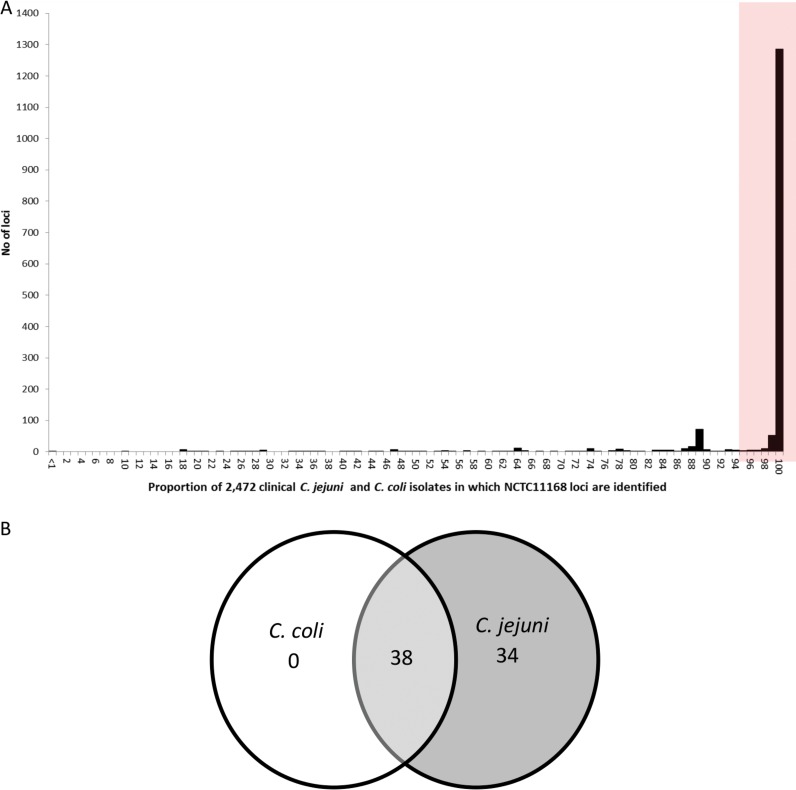
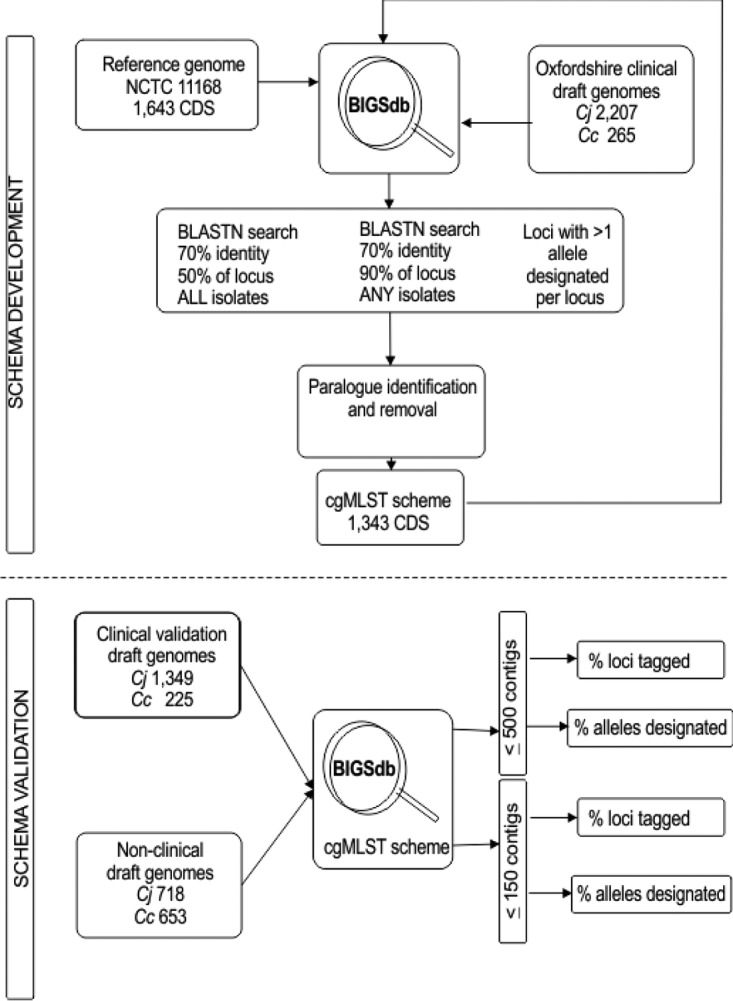
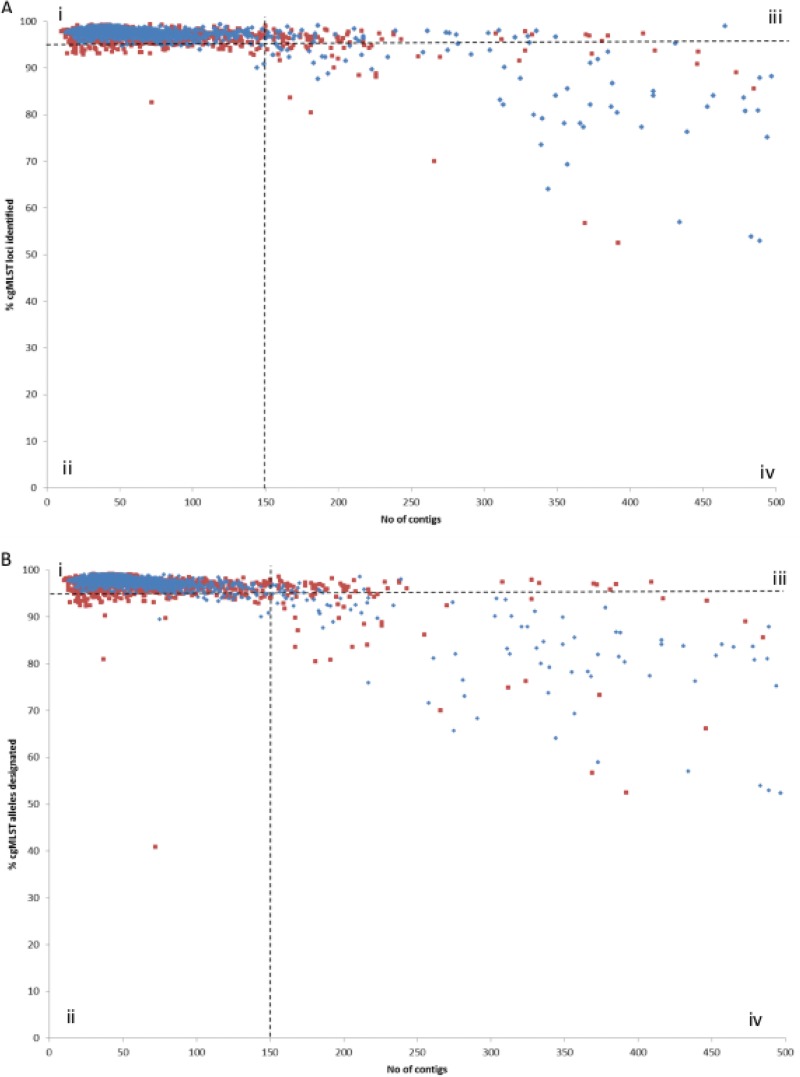
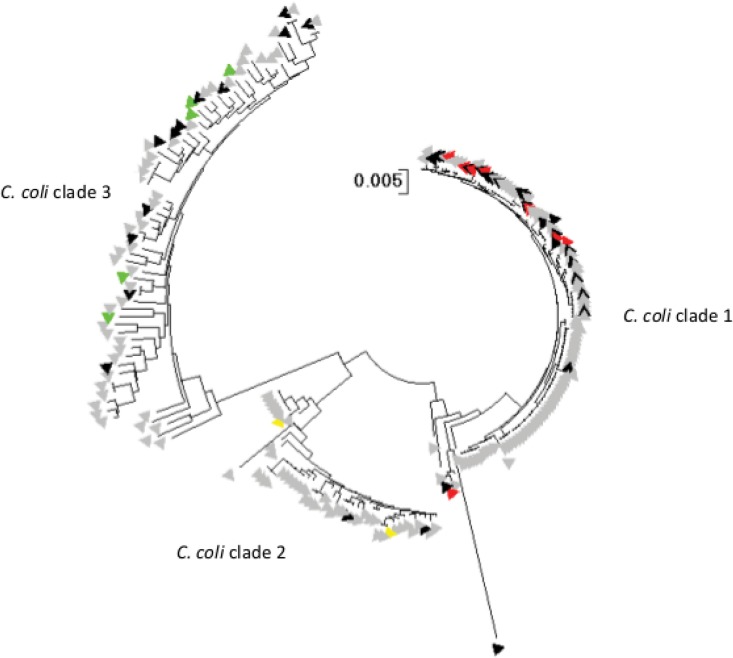
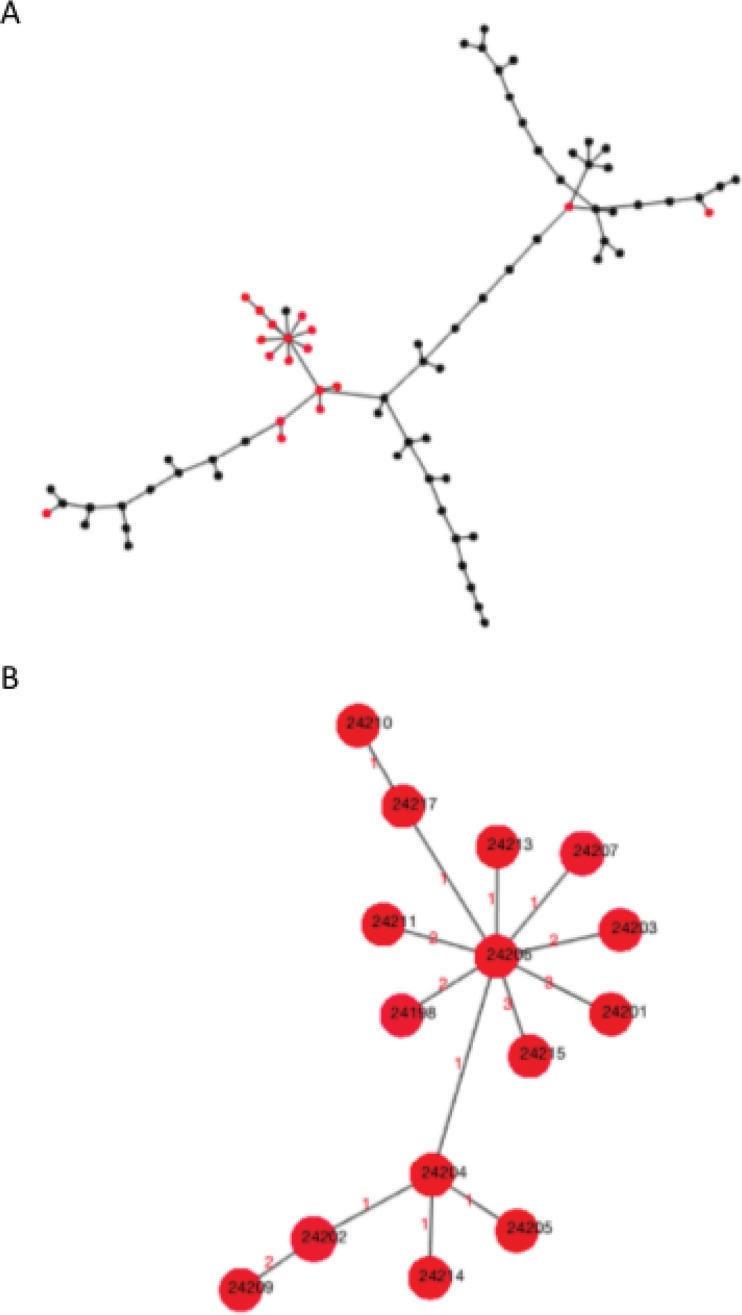
Human campylobacteriosis, caused by and , remains a leading cause of bacterial gastroenteritis in many countries, but the epidemiology of campylobacteriosis outbreaks remains poorly defined, largely due to limitations in the resolution and comparability of isolate characterization methods. Whole-genome sequencing (WGS) data enable the improvement of sequence-based typing approaches, such as multilocus sequence typing (MLST), by substantially increasing the number of loci examined. A core genome MLST (cgMLST) scheme defines a comprehensive set of those loci present in most members of a bacterial group, balancing very high resolution with comparability across the diversity of the group. Here we propose a set of 1,343 loci as a human campylobacteriosis cgMLST scheme (v1.0), the allelic profiles of which can be assigned to core genome sequence types. The 1,343 loci chosen were a subset of the 1,643 loci identified in the reannotation of the genome sequence of isolate NCTC 11168, chosen as being present in >95% of draft genomes of 2,472 representative United Kingdom campylobacteriosis isolates, comprising 2,207 (89.3%) isolates and 265 (10.7%) isolates. Validation of the cgMLST scheme was undertaken with 1,478 further high-quality draft genomes, containing 150 or fewer contiguous sequences, from disease isolate collections: 99.5% of these isolates contained ≥95% of the 1,343 cgMLST loci. In addition to the rapid and effective high-resolution analysis of large numbers of diverse isolates, the cgMLST scheme enabled the efficient identification of very closely related isolates from a well-defined single-source campylobacteriosis outbreak.
由空肠弯曲菌和结肠弯曲菌引起的人类弯曲菌病在许多国家仍是细菌性肠胃炎的主要病因,但弯曲菌病暴发的流行病学仍未得到很好的界定,这主要是由于分离株鉴定方法在分辨率和可比性方面存在局限性。全基因组测序(WGS)数据通过大幅增加所检测位点的数量,能够改进基于序列的分型方法,如多位点序列分型(MLST)。核心基因组MLST(cgMLST)方案定义了一组存在于细菌群体大多数成员中的综合位点,在极高分辨率与群体多样性的可比性之间取得平衡。在此,我们提出一组1343个位点作为人类弯曲菌病cgMLST方案(v1.0),其等位基因谱可被指定为核心基因组序列类型。所选的1343个位点是在空肠弯曲菌分离株NCTC 11168基因组序列重新注释中鉴定出的1643个位点的一个子集,这些位点在2472株英国代表性弯曲菌病分离株的草图基因组中出现率>95%,其中包括2207株(89.3%)空肠弯曲菌分离株和265株(10.7%)结肠弯曲菌分离株。使用来自疾病分离株集合的另外1478个高质量草图基因组(包含150个或更少连续序列)对cgMLST方案进行了验证:这些分离株中有99.5%包含1343个cgMLST位点中的≥95%。除了对大量不同分离株进行快速有效的高分辨率分析外,cgMLST方案还能从明确的单源弯曲菌病暴发中高效鉴定出非常密切相关的分离株。