Johnston Dayle, Earley Bernadette, Cormican Paul, Murray Gerard, Kenny David Anthony, Waters Sinead Mary, McGee Mark, Kelly Alan Kieran, McCabe Matthew Sean
Animal and Bioscience Research Department, Animal & Grassland Research and Innovation Centre, Teagasc Grange, Dunsany, Co. Meath, Ireland.
School of Agriculture Food Science and Veterinary Medicine, University College Dublin, Dublin, Belfield, Dublin 4, Ireland.
BMC Vet Res. 2017 May 2;13(1):118. doi: 10.1186/s12917-017-1035-2.
Bovine respiratory disease (BRD) is caused by growth of single or multiple species of pathogenic bacteria in lung tissue following stress and/or viral infection. Next generation sequencing of 16S ribosomal RNA gene PCR amplicons (NGS 16S amplicon analysis) is a powerful culture-independent open reference method that has recently been used to increase understanding of BRD-associated bacteria in the upper respiratory tract of BRD cattle. However, it has not yet been used to examine the microbiome of the bovine lower respiratory tract. The objective of this study was to use NGS 16S amplicon analysis to identify bacteria in post-mortem lung and lymph node tissue samples harvested from fatal BRD cases and clinically healthy animals. Cranial lobe and corresponding mediastinal lymph node post-mortem tissue samples were collected from calves diagnosed as BRD cases by veterinary laboratory pathologists and from clinically healthy calves. NGS 16S amplicon libraries, targeting the V3-V4 region of the bacterial 16S rRNA gene were prepared and sequenced on an Illumina MiSeq. Quantitative insights into microbial ecology (QIIME) was used to determine operational taxonomic units (OTUs) which corresponded to the 16S rRNA gene sequences.
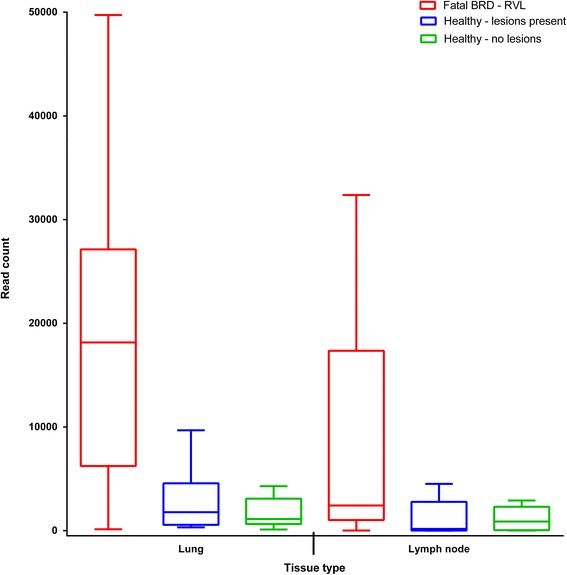
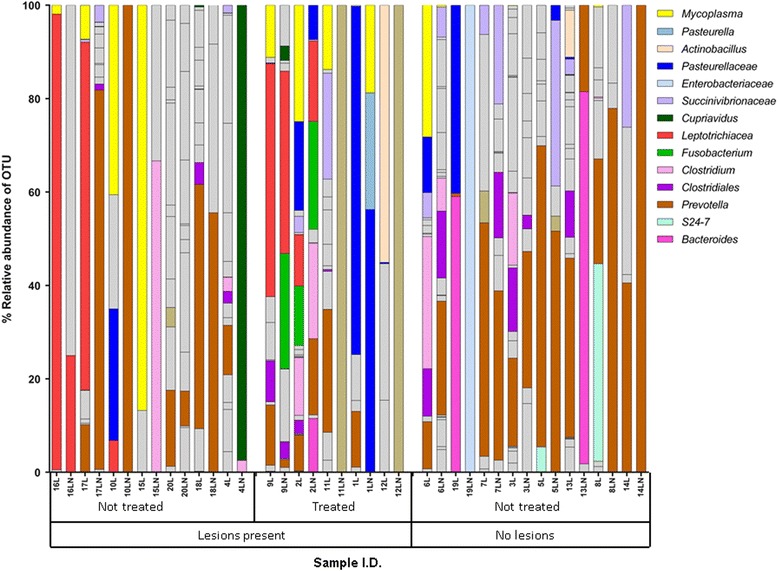
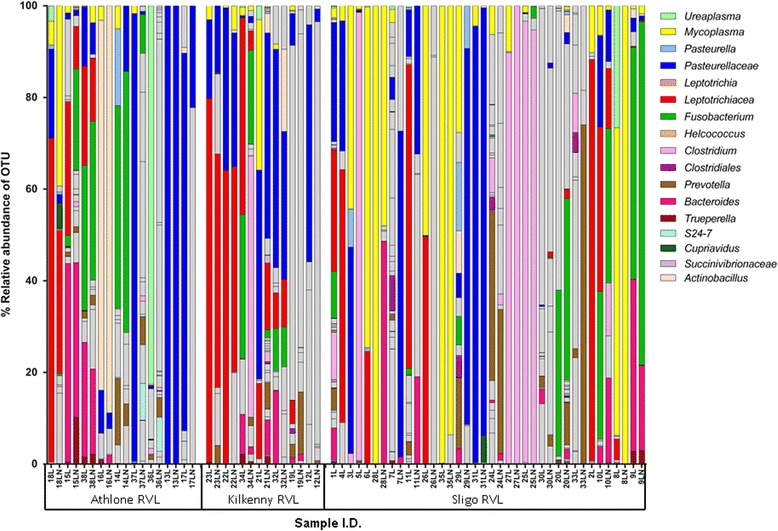
Leptotrichiaceae, Mycoplasma, Pasteurellaceae, and Fusobacterium were the most abundant OTUs identified in the lungs and lymph nodes of the calves which died from BRD. Leptotrichiaceae, Fusobacterium, Mycoplasma, Trueperella and Bacteroides had greater relative abundances in post-mortem lung samples collected from fatal cases of BRD in dairy calves, compared with clinically healthy calves without lung lesions. Leptotrichiaceae, Mycoplasma and Pasteurellaceae showed higher relative abundances in post-mortem lymph node samples collected from fatal cases of BRD in dairy calves, compared with clinically healthy calves without lung lesions. Two Leptotrichiaceae sequence contigs were subsequently assembled from bacterial DNA-enriched shotgun sequences.
The microbiomes of the cranial lung lobe and mediastinal lymph node from calves which died from BRD and from clinically healthy H-F calves have been characterised. Contigs corresponding to the abundant Leptotrichiaceae OTU were sequenced and found not to be identical to any known bacterial genus. This suggests that we have identified a novel bacterial species associated with BRD.
牛呼吸道疾病(BRD)是由应激和/或病毒感染后肺组织中单一或多种病原菌生长引起的。16S核糖体RNA基因PCR扩增子的下一代测序(NGS 16S扩增子分析)是一种强大的非培养依赖型开放参考方法,最近已被用于增进对BRD牛上呼吸道中与BRD相关细菌的了解。然而,它尚未用于检测牛下呼吸道的微生物群。本研究的目的是使用NGS 16S扩增子分析来鉴定从致命BRD病例和临床健康动物采集的死后肺和淋巴结组织样本中的细菌。从经兽医实验室病理学家诊断为BRD病例的犊牛和临床健康的犊牛中采集颅叶和相应的纵隔淋巴结死后组织样本。制备靶向细菌16S rRNA基因V3-V4区域的NGS 16S扩增子文库,并在Illumina MiSeq上进行测序。使用微生物生态学定量见解(QIIME)来确定与16S rRNA基因序列相对应的操作分类单元(OTU)。
在死于BRD的犊牛的肺和淋巴结中鉴定出的最丰富的OTU是纤毛菌科、支原体、巴斯德菌科和梭杆菌。与无肺部病变的临床健康犊牛相比,在从患有BRD的乳用犊牛致命病例采集的死后肺样本中,纤毛菌科、梭杆菌、支原体、特鲁珀菌属和拟杆菌属具有更高的相对丰度。与无肺部病变的临床健康犊牛相比,在从患有BRD的乳用犊牛致命病例采集的死后淋巴结样本中,纤毛菌科、支原体和巴斯德菌科显示出更高的相对丰度。随后从富含细菌DNA的鸟枪法序列中组装出两个纤毛菌科序列重叠群。
已对死于BRD的犊牛和临床健康的H-F犊牛的颅肺叶和纵隔淋巴结的微生物群进行了表征。对与丰富的纤毛菌科OTU相对应的重叠群进行了测序,发现与任何已知细菌属都不相同。这表明我们已经鉴定出一种与BRD相关的新型细菌物种。