Department of Veterinary Sciences, Clinical section, University of Turin, Largo Paolo Braccini 2, 10095, Grugliasco, TO, Italy.
Istituto Zooprofilattico Sperimentale del Piemonte, Liguria e Valle d'Aosta, Via Bologna 148, 10154, Turin, TO, Italy.
Microbiome. 2017 Nov 21;5(1):152. doi: 10.1186/s40168-017-0372-5.
The microbiota of the bovine upper respiratory tract has been recently characterized, but no data for the lower respiratory tract are available. A major health problem in bovine medicine is infectious bronchopneumonia, the most common respiratory syndrome affecting cattle. With this study, we used 16S rRNA gene sequencing to characterize and compare the microbial community composition of the upper and lower respiratory tracts in calves.
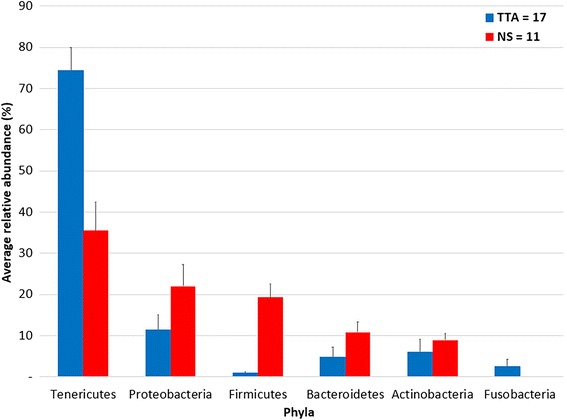
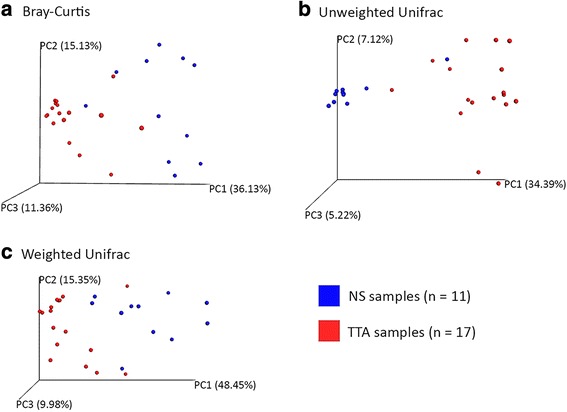
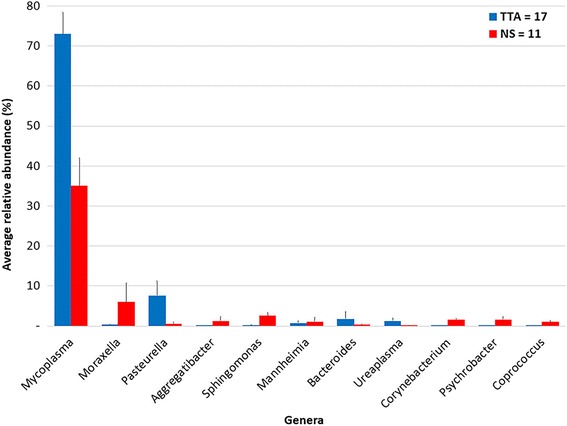
The microbiota of the upper (nasal swab [NS]) and the lower (trans-tracheal aspiration [TTA]) respiratory tracts of 19 post-weaned Piedmontese calves with (8/19) and without (11/19) clinical signs of respiratory disease, coming from six different farms, was characterized by 16S rRNA gene metabarcoding. A total of 29 phyla (29 in NS, 21 in TTA) and 305 genera (289 in NS, 182 in TTA) were identified. Mycoplasma (60.8%) was the most abundant genus identified in both the NS (27.3%) and TTA (76.7%) samples, followed by Moraxella (16.6%) in the NS and Pasteurella (7.3%) in the TTA samples. Pasteurella multocida (7.3% of total operational taxonomic units [OTUs]) was the most abundant species in the TTA and Psychrobacter sanguinis (1.1% of total OTUs) in the NS samples. Statistically significant differences between the NS and the TTA samples were found for both alpha (Shannon index, observed species, Chao1 index, and Simpson index; P = 0.001) and beta (Adonis; P = 0.001) diversity. Comparison of the NS and TTA samples by farm origin and clinical signs revealed no statistical difference (P > 0.05), except for farm origin for the NS samples when compared by the unweighted UniFrac metric (P = 0.05).
Using 16S rRNA gene sequencing, we characterized the microbiota of the upper and lower respiratory tracts of calves, both healthy individuals and those with clinical signs of respiratory disease. Our results suggest that environmental factors may influence the composition of the upper airway microbiota in cattle. While the two microbial communities (upper and lower airways) differed in microbial composition, they shared several OTUs, suggesting that the lung microbiota may be a self-sustaining, more homogeneous ecosystem, influenced by the upper respiratory tract microbiota.
牛上呼吸道的微生物群已被最近描述,但下呼吸道的微生物群尚未可知。牛医学中的一个主要健康问题是传染性支气管肺炎,这是一种影响牛的最常见呼吸道综合征。在这项研究中,我们使用 16S rRNA 基因测序来描述和比较犊牛上呼吸道和下呼吸道的微生物群落组成。
19 头断奶后皮埃蒙特牛的上呼吸道(鼻拭子 [NS])和下呼吸道(经气管抽吸 [TTA])的微生物群,来自六个不同农场,有(8/19)和没有(11/19)临床呼吸道疾病症状,通过 16S rRNA 基因 metabarcoding 进行了描述。共鉴定出 29 个门(NS 中有 29 个,TTA 中有 21 个)和 305 个属(NS 中有 289 个,TTA 中有 182 个)。支原体(60.8%)是 NS(27.3%)和 TTA(76.7%)样本中最丰富的属,其次是 NS 中的莫拉菌(16.6%)和 TTA 中的巴氏杆菌(7.3%)。在 TTA 中最丰富的物种是多杀巴斯德菌(总分类单元 [OTUs] 的 7.3%),而在 NS 中最丰富的物种是血链球菌(总 OTUs 的 1.1%)。在 NS 和 TTA 样本之间,无论是在 alpha 多样性(Shannon 指数、观察到的物种、Chao1 指数和 Simpson 指数;P = 0.001)还是 beta 多样性(Adonis;P = 0.001)方面都存在显著差异。通过农场来源和临床症状对 NS 和 TTA 样本进行比较,除了 NS 样本的农场来源在非加权 UniFrac 度量方面存在统计学差异(P = 0.05)外,没有发现统计学差异(P > 0.05)。
本研究使用 16S rRNA 基因测序,描述了健康犊牛和有临床呼吸道疾病症状的犊牛上呼吸道和下呼吸道的微生物群。我们的结果表明,环境因素可能影响牛上呼吸道微生物群的组成。虽然这两个微生物群落(上呼吸道和下呼吸道)在微生物组成上存在差异,但它们共享了几个 OTUs,这表明肺部微生物群可能是一个自我维持的、更均匀的生态系统,受上呼吸道微生物群的影响。