School of Mathematics and Statistics, Hainan Normal University, Haikou, 570100, P. R. China.
College of Life Sciences, Zhejiang Sci-Tech University, Hangzhou, 310018, P. R. China.
Sci Rep. 2017 May 8;7(1):1545. doi: 10.1038/s41598-017-01699-z.
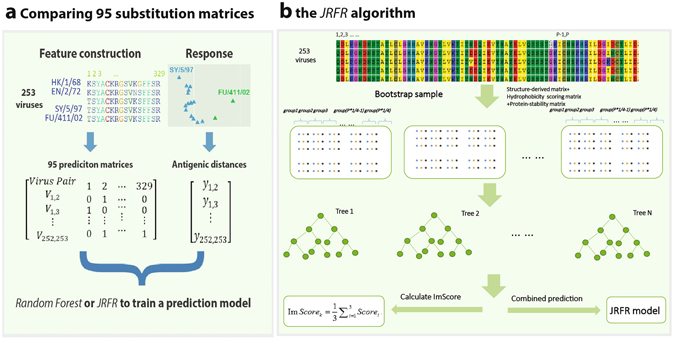
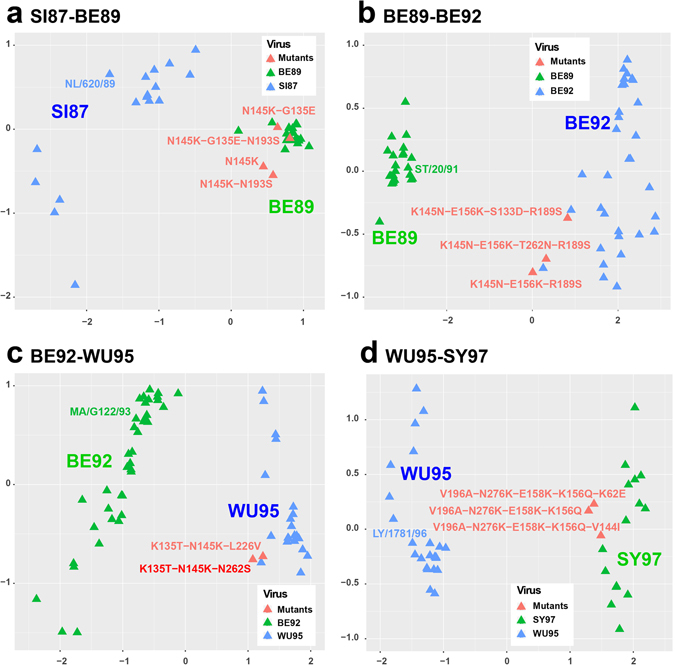
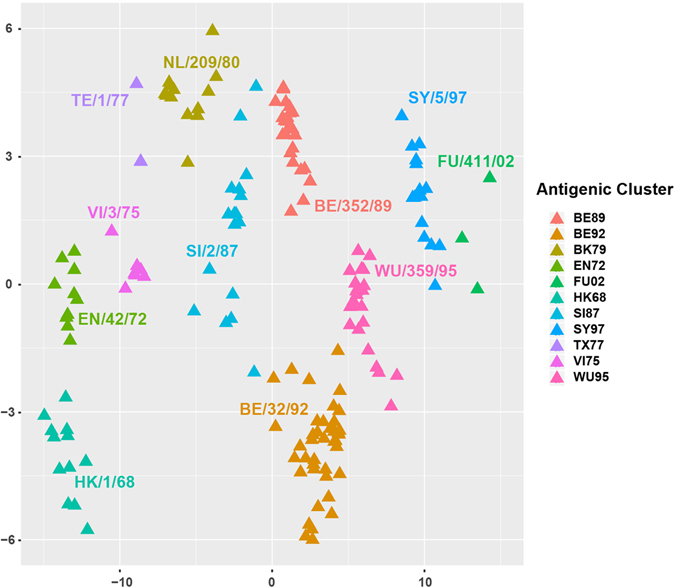
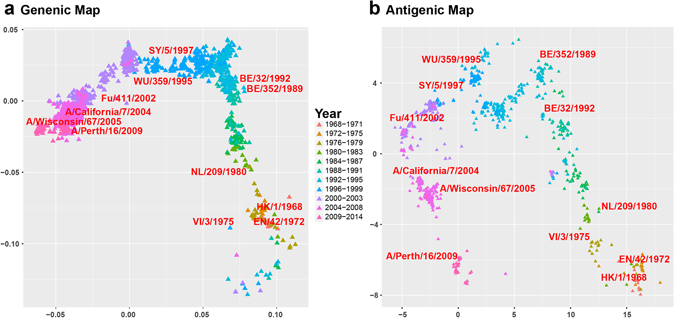
Timely identification of emerging antigenic variants is critical to influenza vaccine design. The accuracy of a sequence-based antigenic prediction method relies on the choice of amino acids substitution matrices. In this study, we first compared a comprehensive 95 substitution matrices reflecting various amino acids properties in predicting the antigenicity of influenza viruses by a random forest model. We then proposed a novel algorithm called joint random forest regression (JRFR) to jointly consider top substitution matrices. We applied JRFR to human H3N2 seasonal influenza data from 1968 to 2003. A 10-fold cross-validation shows that JRFR outperforms other popular methods in predicting antigenic variants. In addition, our results suggest that structure features are most relevant to influenza antigenicity. By restricting the analysis to data involving two adjacent antigenic clusters, we inferred a few key amino acids mutation driving the 11 historical antigenic drift events, pointing to experimentally validated mutations. Finally, we constructed an antigenic cartography of all H3N2 viruses with hemagglutinin (the glycoprotein on the surface of the influenza virus responsible for its binding to host cells) sequence available from NCBI flu database, and showed an overall correspondence and local inconsistency between genetic and antigenic evolution of H3N2 influenza viruses.
及时识别新出现的抗原变异体对于流感疫苗设计至关重要。基于序列的抗原预测方法的准确性依赖于氨基酸替代矩阵的选择。在本研究中,我们首先通过随机森林模型比较了综合的 95 个反映各种氨基酸特性的替代矩阵在预测流感病毒抗原性方面的表现。然后,我们提出了一种称为联合随机森林回归(JRFR)的新算法,用于联合考虑顶级替代矩阵。我们将 JRFR 应用于 1968 年至 2003 年期间的人类 H3N2 季节性流感数据。10 倍交叉验证表明,JRFR 在预测抗原变体方面优于其他流行方法。此外,我们的结果表明,结构特征与流感抗原性最相关。通过将分析限制在涉及两个相邻抗原簇的数据中,我们推断出了几个关键的氨基酸突变,这些突变驱动了 11 次历史抗原漂移事件,指向了经过实验验证的突变。最后,我们构建了一个来自 NCBI flu 数据库的所有 H3N2 病毒血凝素(流感病毒表面负责与宿主细胞结合的糖蛋白)序列的抗原图谱,并显示了 H3N2 流感病毒遗传和抗原进化之间的整体一致性和局部不一致性。