Martin Constance J, Cadena Anthony M, Leung Vivian W, Lin Philana Ling, Maiello Pauline, Hicks Nathan, Chase Michael R, Flynn JoAnne L, Fortune Sarah M
Department of Immunology and Infectious Diseases, Harvard T. H. Chan School of Public Health, Boston, Massachusetts, USA.
Ragon Institute of Massachusetts General Hospital, Massachusetts Institute of Technology and Harvard, Cambridge, Massachusetts, USA.
mBio. 2017 May 9;8(3):e00312-17. doi: 10.1128/mBio.00312-17.
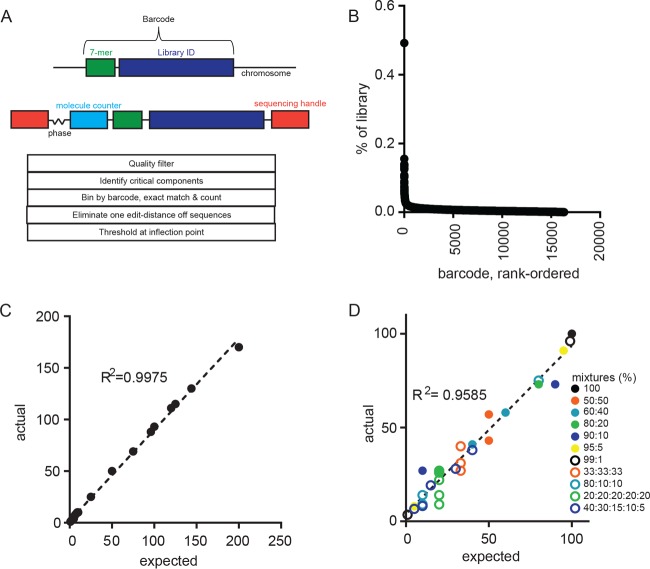
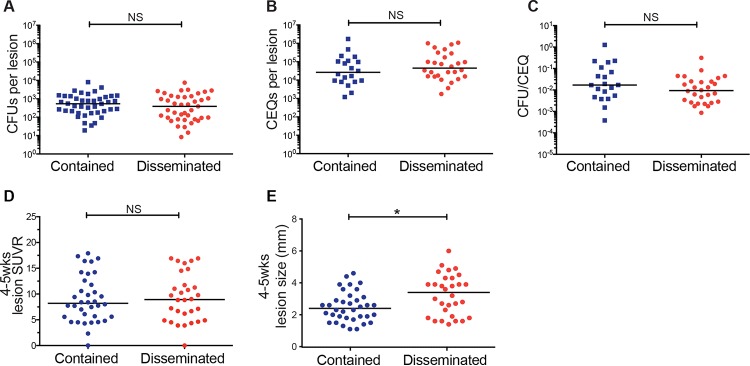
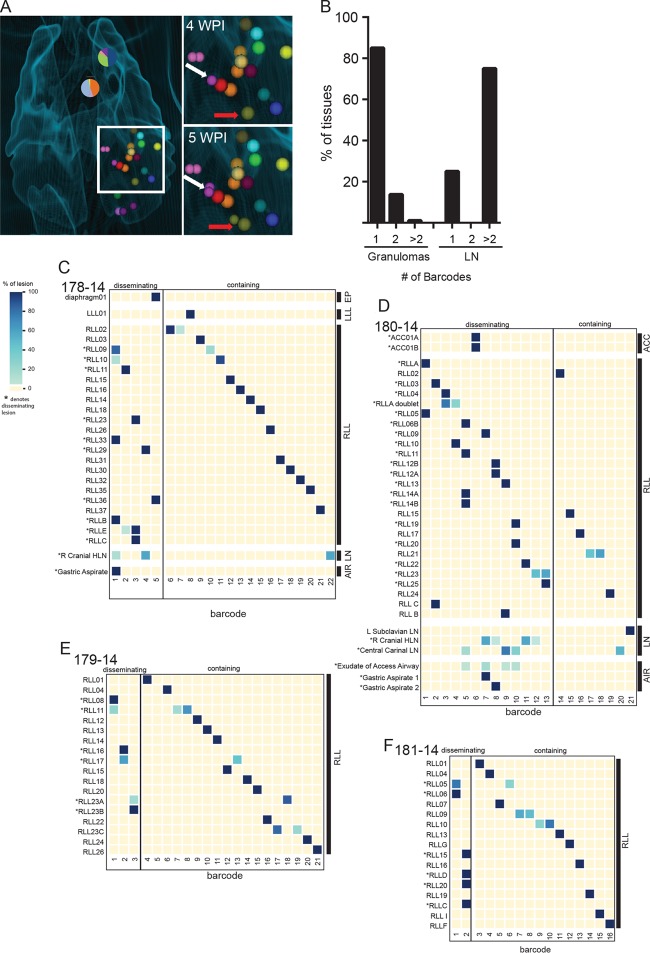
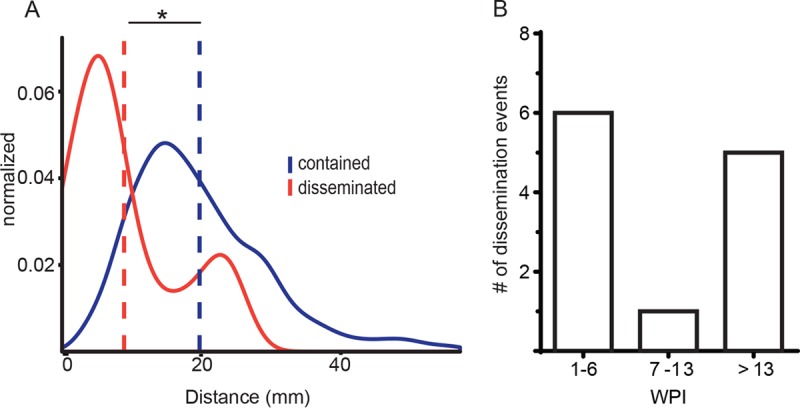
Infection with causes a spectrum of outcomes; the majority of individuals contain but do not eliminate the infection, while a small subset present with primary active tuberculosis (TB) disease. This variability in infection outcomes is recapitulated at the granuloma level within each host, such that some sites of infection can be fully cleared while others progress. Understanding the spectrum of TB outcomes requires new tools to deconstruct the mechanisms underlying differences in granuloma fate. Here, we use novel genome-encoded barcodes to uniquely tag individual bacilli, enabling us to quantitatively track the trajectory of each infecting bacterium in a macaque model of TB. We also introduce a robust bioinformatics pipeline capable of identifying and counting barcode sequences within complex mixtures and at various read depths. By coupling this tagging strategy with serial positron emission tomography coregistered with computed tomography (PET/CT) imaging of lung pathology in macaques, we define a lesional map of infection dynamics. We find that there is no significant infection bottleneck, but there are significant constraints on productive bacterial trafficking out of primary granulomas. Our findings validate our barcoding approach and demonstrate its utility in probing lesion-specific biology and dissemination. This novel technology has the potential to greatly enhance our understanding of local dynamics in tuberculosis. Classically, infection was thought to result in either latent infection or active disease. More recently, the field has recognized that there is a spectrum of infection clinical outcomes. Within a single host, this spectrum is recapitulated at the granuloma level, where there can simultaneously be lesional sterilization and poorly contained disease. To better understand the lesional biology of TB infection, we digitally barcoded to quantitatively track the fate of each infecting bacterium. By combining this technology with serial PET-CT imaging, we can dynamically track both bacterial populations and granuloma trajectories. We demonstrate that there is little constraint on the bacterial population at the time of infection. However, the granuloma imposes a strong bottleneck on dissemination, and the subset of granulomas at risk of dissemination can be distinguished by physical features.
感染结核分枝杆菌会导致一系列结果;大多数个体感染后携带但未清除该感染,而一小部分个体表现为原发性活动性结核病。这种感染结果的变异性在每个宿主体内的肉芽肿水平上也有体现,即一些感染部位可以完全清除,而另一些则会进展。要理解结核病结果的范围,需要新的工具来剖析肉芽肿命运差异背后的机制。在这里,我们使用新型基因组编码条形码对单个结核分枝杆菌进行独特标记,从而能够在猕猴结核病模型中定量追踪每一个感染细菌的轨迹。我们还引入了一个强大的生物信息学流程,能够在复杂混合物中以及不同读取深度下识别和计数条形码序列。通过将这种标记策略与猕猴肺部病理的计算机断层扫描(PET/CT)成像同步的系列正电子发射断层扫描相结合,我们定义了结核分枝杆菌感染动态的病灶图谱。我们发现不存在显著的感染瓶颈,但从原发性肉芽肿中进行有效的细菌传播存在重大限制。我们的研究结果验证了我们的条形码方法,并证明了其在探究病灶特异性生物学和传播方面的效用。这项新技术有可能极大地增进我们对结核病局部动态的理解。传统上,结核分枝杆菌感染被认为要么导致潜伏感染,要么导致活动性疾病。最近,该领域认识到结核分枝杆菌感染存在一系列临床结果。在单个宿主体内,这种范围在肉芽肿水平上也有体现,在那里可以同时存在病灶杀菌和控制不佳的疾病。为了更好地理解结核分枝杆菌感染的病灶生物学,我们对结核分枝杆菌进行数字条形码标记以定量追踪每一个感染细菌的命运。通过将这项技术与系列PET-CT成像相结合,我们可以动态追踪细菌群体和肉芽肿轨迹。我们证明在感染时对细菌群体几乎没有限制。然而,肉芽肿对传播构成了强大的瓶颈,并且有传播风险的肉芽肿子集可以通过物理特征来区分。