Eng Christine L P, Tong Joo Chuan, Tan Tin Wee
Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, 117597 Singapore, Singapore.
Institute of High Performance Computing, A*Star, 138632 Singapore, Singapore.
Int J Mol Sci. 2017 May 25;18(6):1135. doi: 10.3390/ijms18061135.
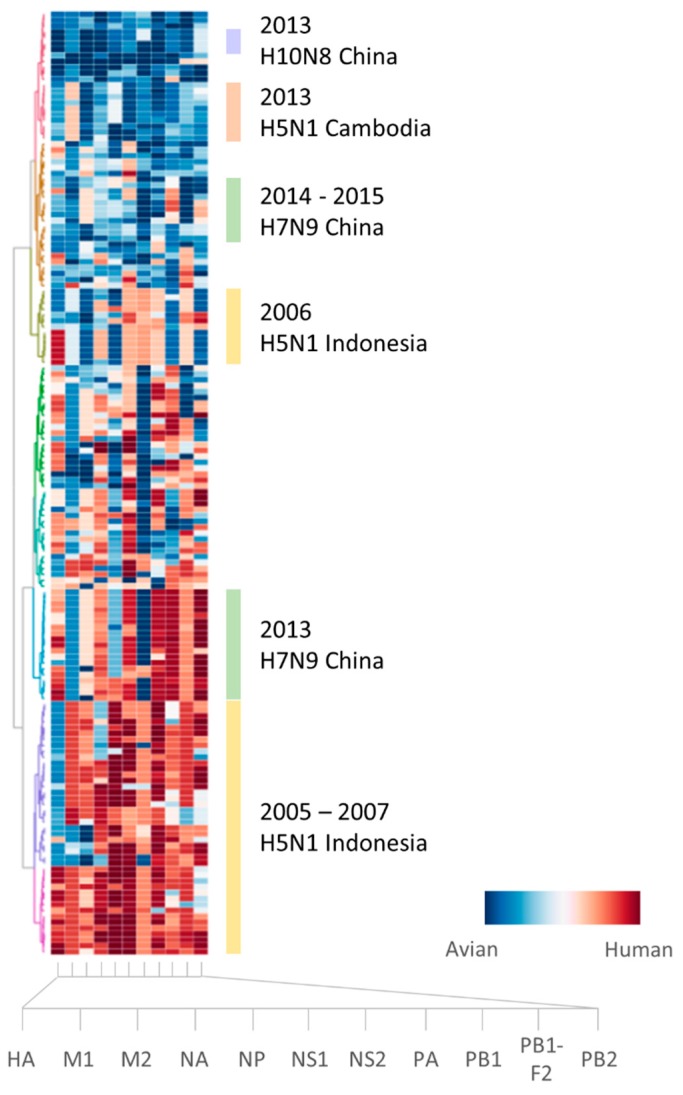
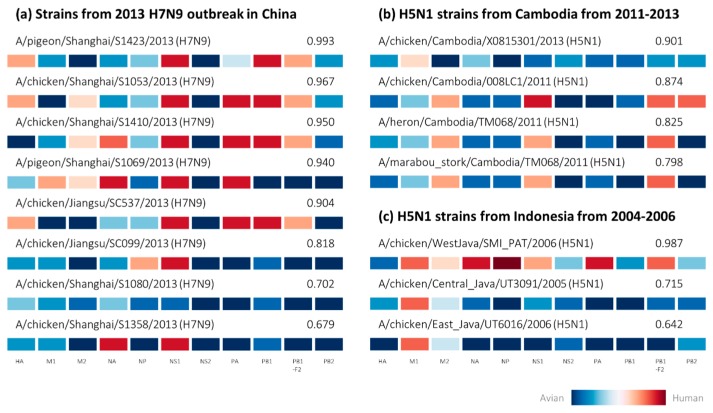
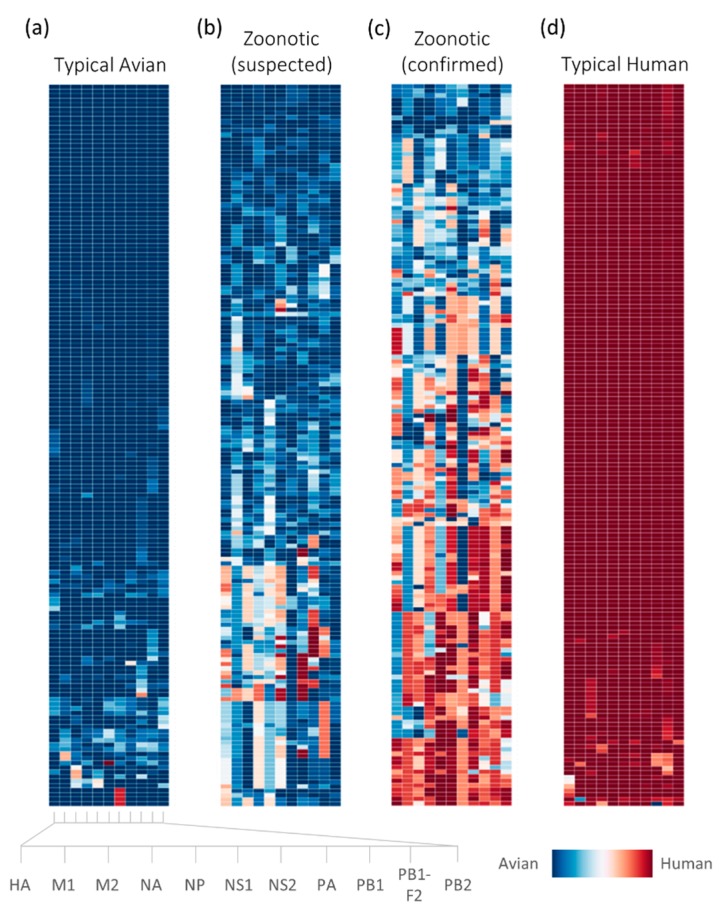
Influenza A viruses remain a significant health problem, especially when a novel subtype emerges from the avian population to cause severe outbreaks in humans. Zoonotic viruses arise from the animal population as a result of mutations and reassortments, giving rise to novel strains with the capability to evade the host species barrier and cause human infections. Despite progress in understanding interspecies transmission of influenza viruses, we are no closer to predicting zoonotic strains that can lead to an outbreak. We have previously discovered distinct host tropism protein signatures of avian, human and zoonotic influenza strains obtained from host tropism predictions on individual protein sequences. Here, we apply machine learning approaches on the signatures to build a computational model capable of predicting zoonotic strains. The zoonotic strain prediction model can classify avian, human or zoonotic strains with high accuracy, as well as providing an estimated zoonotic risk. This would therefore allow us to quickly determine if an influenza virus strain has the potential to be zoonotic using only protein sequences. The swift identification of potential zoonotic strains in the animal population using the zoonotic strain prediction model could provide us with an early indication of an imminent influenza outbreak.
甲型流感病毒仍然是一个重大的健康问题,尤其是当一种新型亚型从禽类群体中出现并在人类中引发严重疫情时。人畜共患病毒是由于突变和重配从动物群体中产生的,从而产生了能够突破宿主物种屏障并导致人类感染的新型毒株。尽管在理解流感病毒的跨物种传播方面取得了进展,但我们距离预测可能导致疫情爆发的人畜共患毒株仍没有更近一步。我们之前通过对单个蛋白质序列进行宿主嗜性预测,发现了禽类、人类和人畜共患流感毒株不同的宿主嗜性蛋白特征。在此,我们对这些特征应用机器学习方法来构建一个能够预测人畜共患毒株的计算模型。人畜共患毒株预测模型能够高精度地对禽类、人类或人畜共患毒株进行分类,并提供估计的人畜共患风险。因此,这将使我们仅使用蛋白质序列就能快速确定一种流感病毒毒株是否具有人畜共患的潜力。使用人畜共患毒株预测模型在动物群体中快速识别潜在的人畜共患毒株,能够为我们提供即将发生流感疫情的早期迹象。