Department of Biomedical Engineering, University of Texas at Austin, Austin, Texas, 78712.
Department of Chemistry, Washington University in St. Louis, St. Louis, Missouri, 63130.
J Comput Chem. 2017 Sep 5;38(23):2047-2055. doi: 10.1002/jcc.24853. Epub 2017 Jun 10.
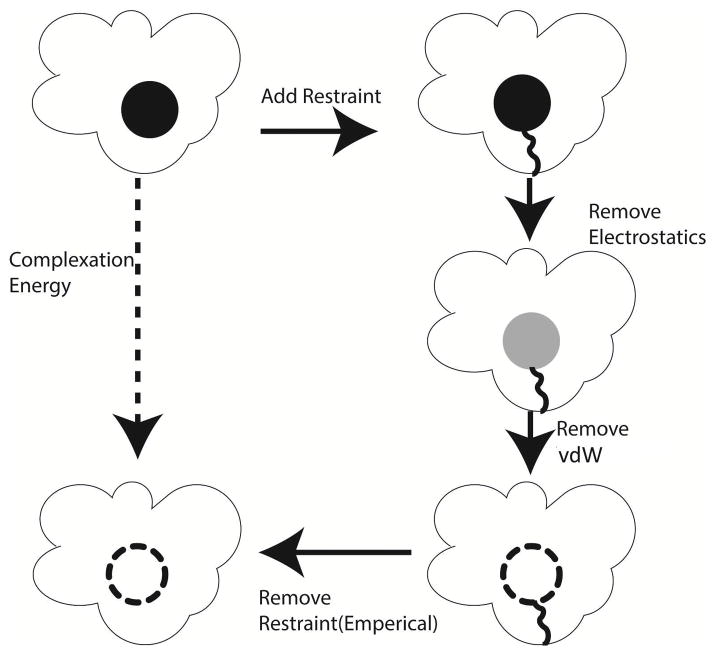
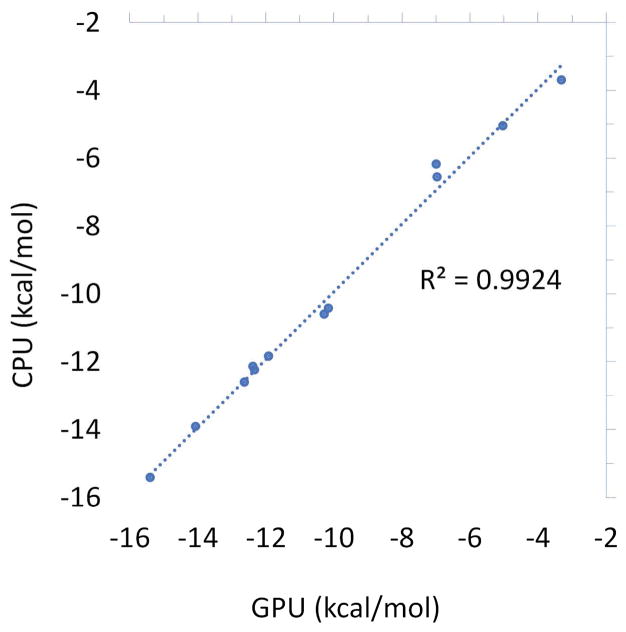
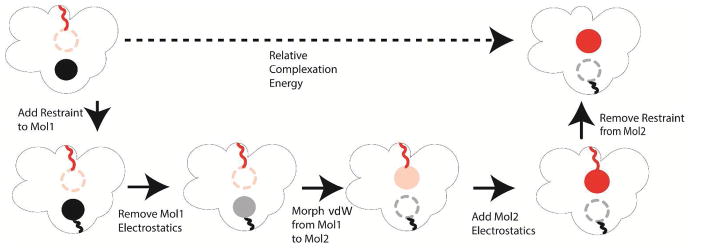
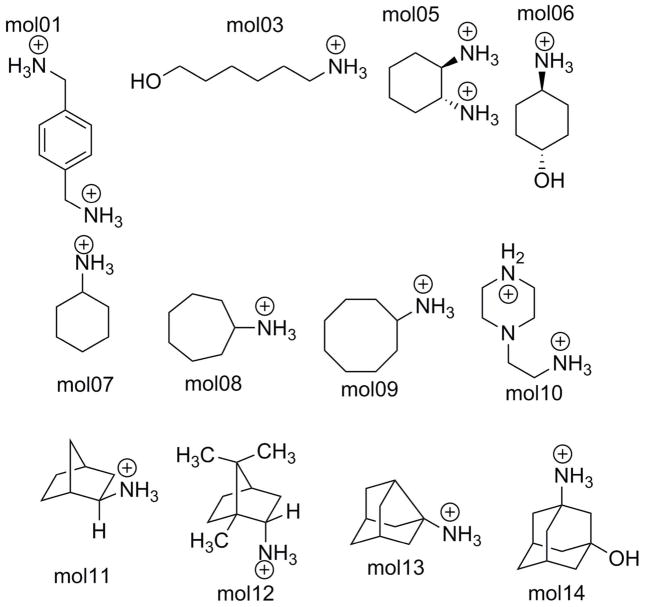
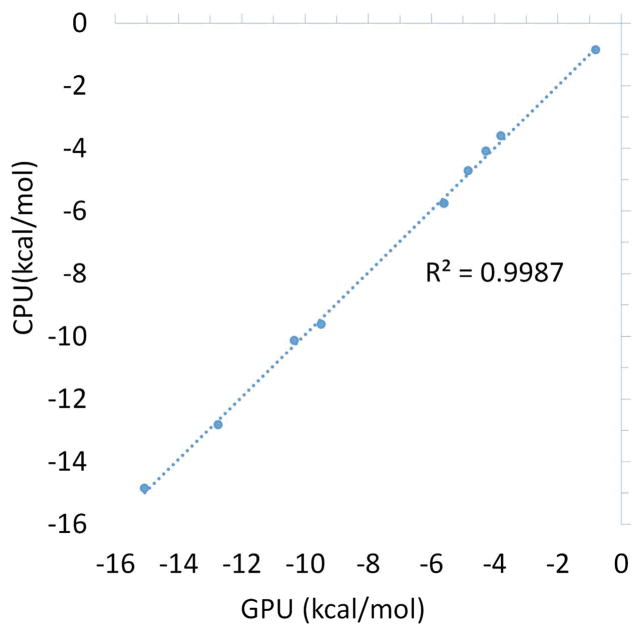
The capabilities of the polarizable force fields for alchemical free energy calculations have been limited by the high computational cost and complexity of the underlying potential energy functions. In this work, we present a GPU-based general alchemical free energy simulation platform for polarizable potential AMOEBA. Tinker-OpenMM, the OpenMM implementation of the AMOEBA simulation engine has been modified to enable both absolute and relative alchemical simulations on GPUs, which leads to a ∼200-fold improvement in simulation speed over a single CPU core. We show that free energy values calculated using this platform agree with the results of Tinker simulations for the hydration of organic compounds and binding of host-guest systems within the statistical errors. In addition to absolute binding, we designed a relative alchemical approach for computing relative binding affinities of ligands to the same host, where a special path was applied to avoid numerical instability due to polarization between the different ligands that bind to the same site. This scheme is general and does not require ligands to have similar scaffolds. We show that relative hydration and binding free energy calculated using this approach match those computed from the absolute free energy approach. © 2017 Wiley Periodicals, Inc.
极化力场在计算炼金术自由能方面的能力受到潜在能量函数计算成本高和复杂的限制。在这项工作中,我们提出了一个基于 GPU 的极化势 AMOEBA 通用炼金术自由能模拟平台。Tinker-OpenMM 是 AMOEBA 模拟引擎的 OpenMM 实现,已经过修改,可在 GPU 上进行绝对和相对炼金术模拟,这使得模拟速度相对于单个 CPU 核心提高了约 200 倍。我们表明,使用该平台计算的自由能值与 Tinker 模拟有机化合物水合和主体-客体体系结合的结果在统计误差范围内一致。除了绝对结合,我们还设计了一种相对炼金术方法来计算配体与同一主体的相对结合亲和力,其中应用了一种特殊路径来避免由于结合到同一位点的不同配体之间的极化引起的数值不稳定性。该方案是通用的,不需要配体具有相似的支架。我们表明,使用该方法计算的相对水合和结合自由能与从绝对自由能方法计算得到的结果匹配。© 2017 威利父子公司