Jin Yumi, Schäffer Alejandro A, Sherry Stephen T, Feolo Michael
National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, United States of America.
PLoS One. 2017 Jun 13;12(6):e0179106. doi: 10.1371/journal.pone.0179106. eCollection 2017.
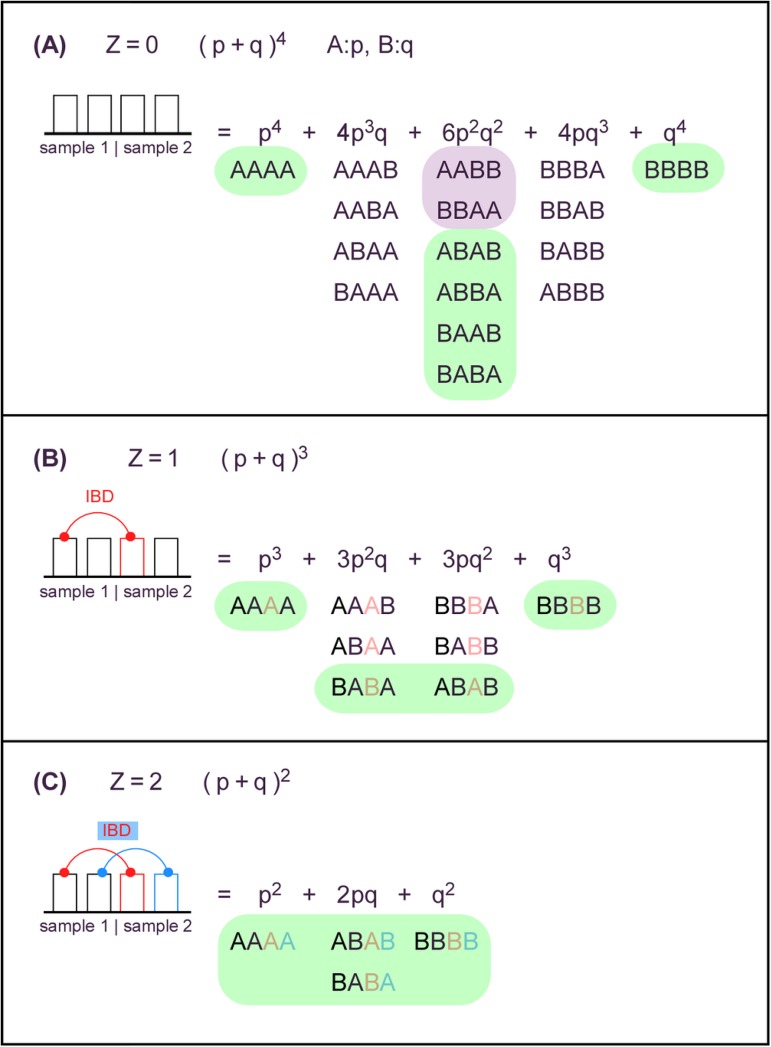
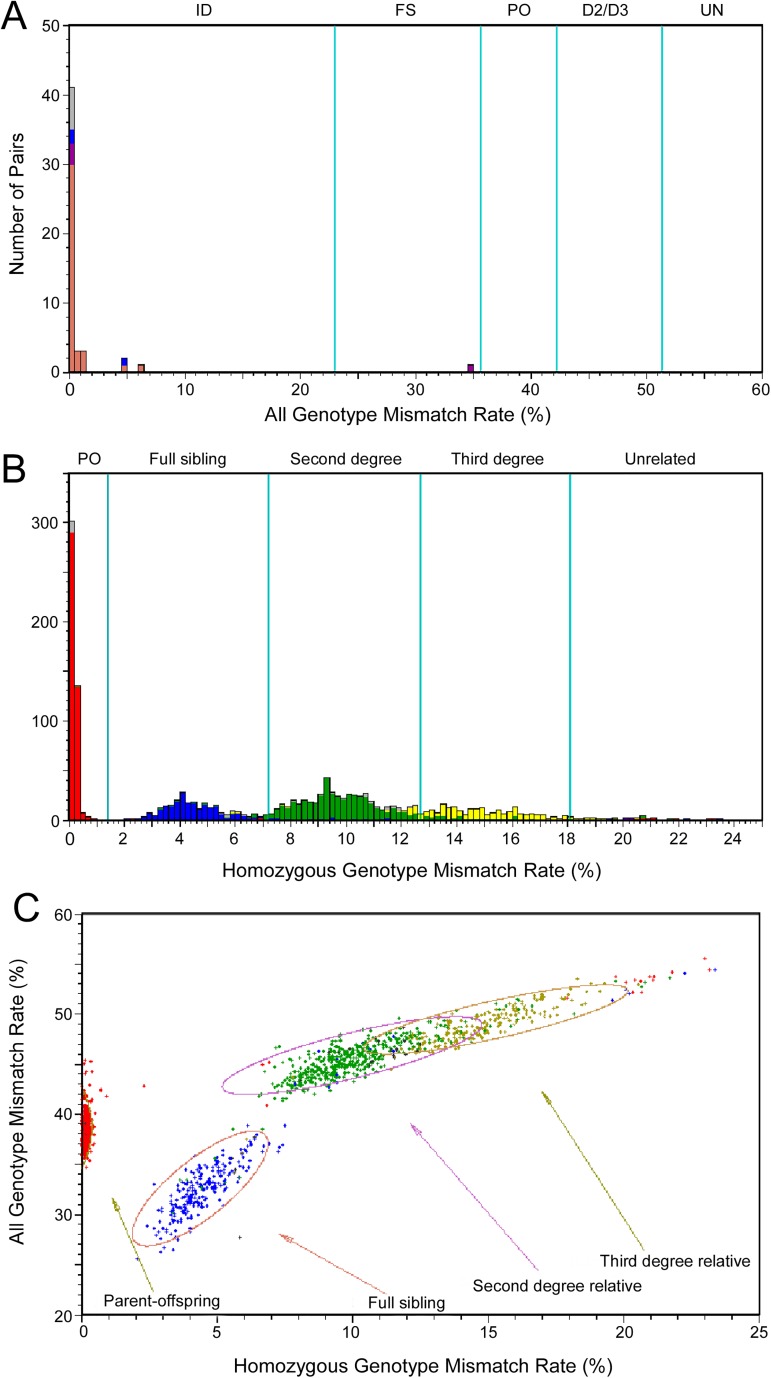
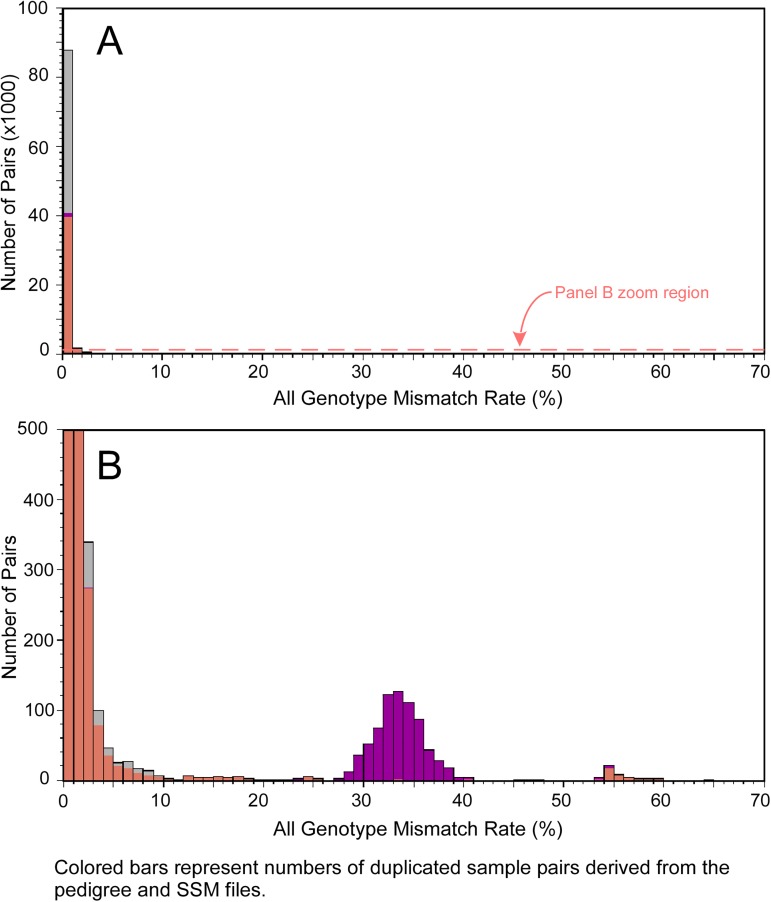
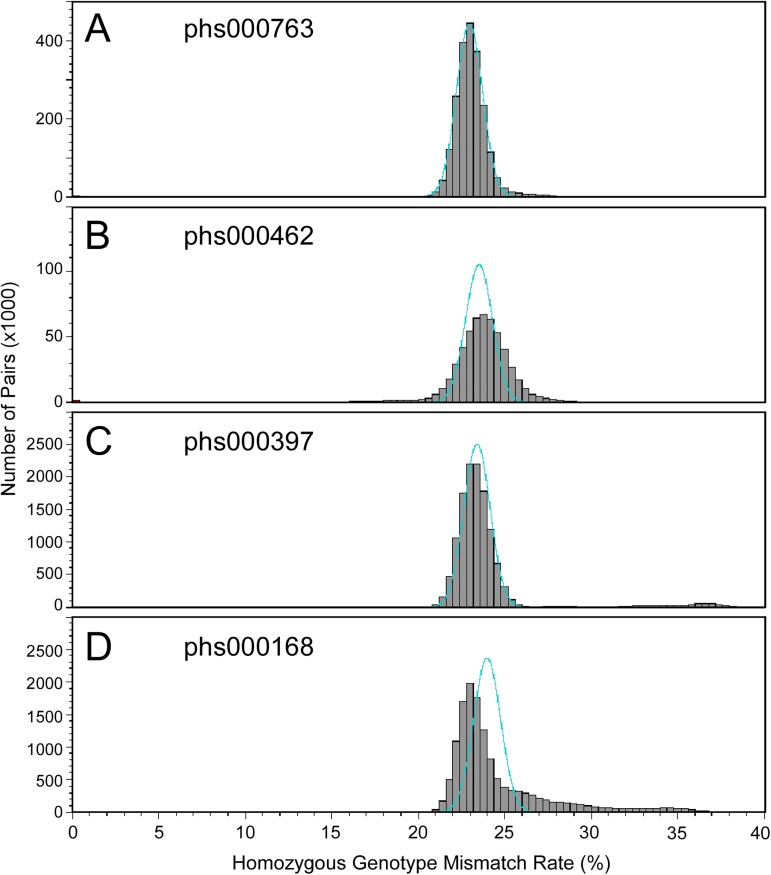
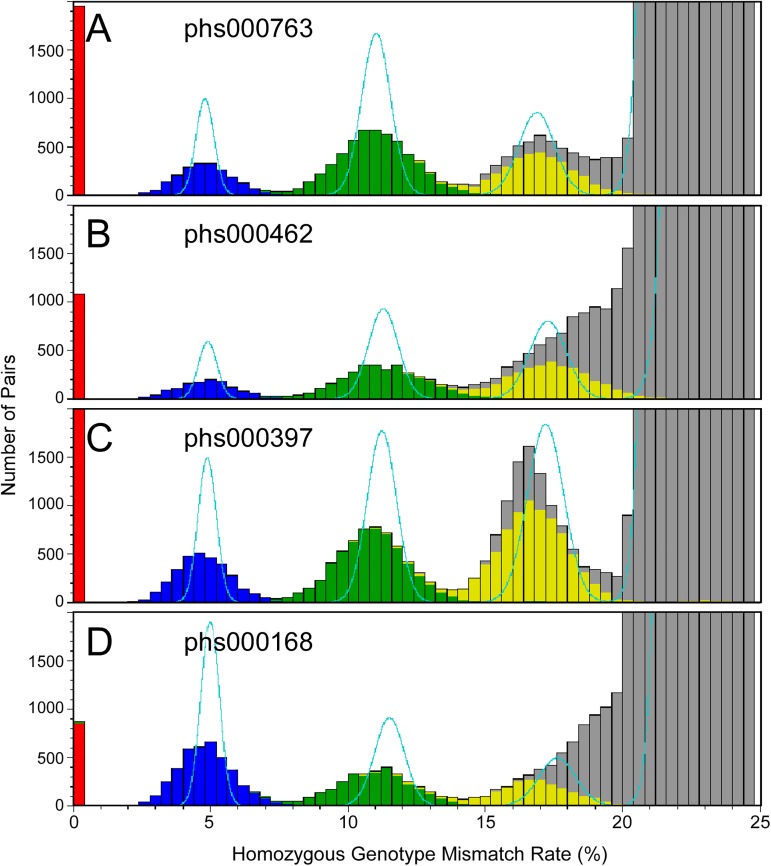
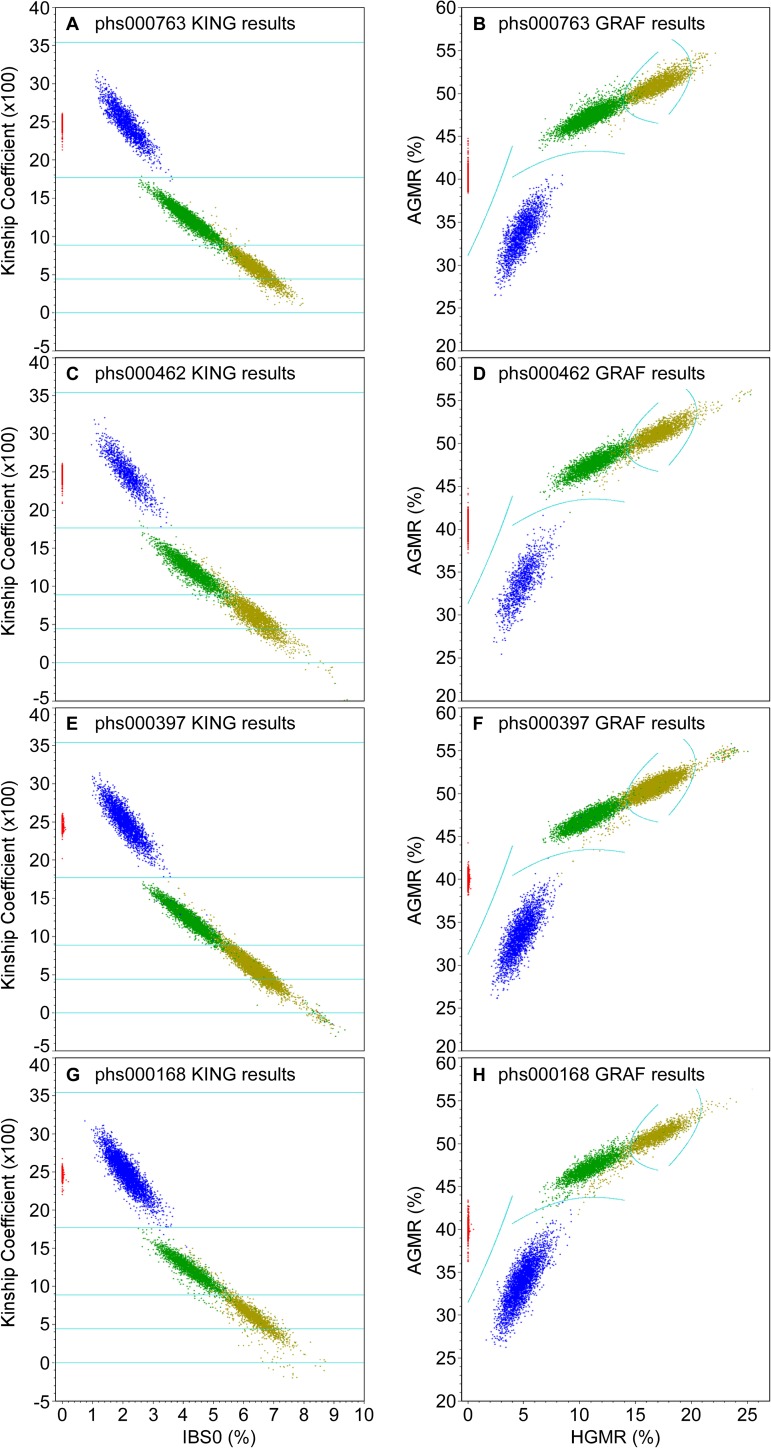
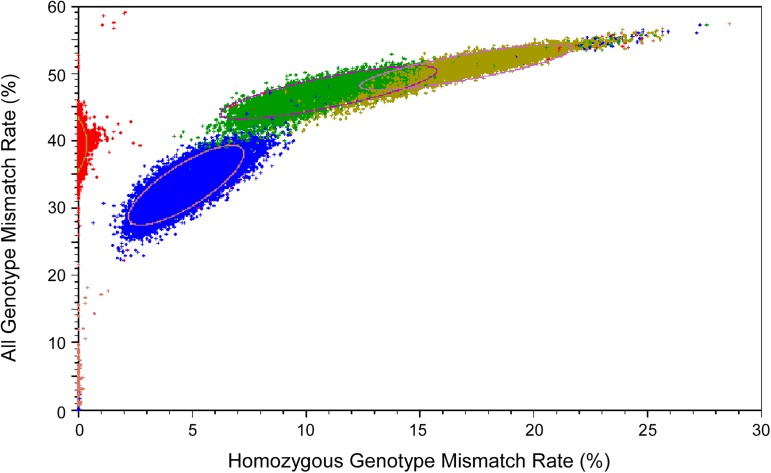
Genome-wide association studies (GWAS) usually rely on the assumption that different samples are not from closely related individuals. Detection of duplicates and close relatives becomes more difficult both statistically and computationally when one wants to combine datasets that may have been genotyped on different platforms. The dbGaP repository at the National Center of Biotechnology Information (NCBI) contains datasets from hundreds of studies with over one million samples. There are many duplicates and closely related individuals both within and across studies from different submitters. Relationships between studies cannot always be identified by the submitters of individual datasets. To aid in curation of dbGaP, we developed a rapid statistical method called Genetic Relationship and Fingerprinting (GRAF) to detect duplicates and closely related samples, even when the sets of genotyped markers differ and the DNA strand orientations are unknown. GRAF extracts genotypes of 10,000 informative and independent SNPs from genotype datasets obtained using different methods, and implements quick algorithms that enable it to find all of the duplicate pairs from more than 880,000 samples within and across dbGaP studies in less than two hours. In addition, GRAF uses two statistical metrics called All Genotype Mismatch Rate (AGMR) and Homozygous Genotype Mismatch Rate (HGMR) to determine subject relationships directly from the observed genotypes, without estimating probabilities of identity by descent (IBD), or kinship coefficients, and compares the predicted relationships with those reported in the pedigree files. We implemented GRAF in a freely available C++ program of the same name. In this paper, we describe the methods in GRAF and validate the usage of GRAF on samples from the dbGaP repository. Other scientists can use GRAF on their own samples and in combination with samples downloaded from dbGaP.
全基因组关联研究(GWAS)通常依赖于不同样本并非来自密切相关个体的假设。当人们想要合并可能在不同平台上进行基因分型的数据集时,无论是在统计上还是计算上,检测重复样本和近亲都变得更加困难。美国国立生物技术信息中心(NCBI)的dbGaP数据库包含来自数百项研究的数据集,样本超过100万个。不同提交者的研究内部和研究之间都存在许多重复样本和密切相关的个体。单个数据集的提交者并不总能识别研究之间的关系。为了协助管理dbGaP,我们开发了一种名为遗传关系与指纹识别(GRAF)的快速统计方法,用于检测重复样本和密切相关的样本,即使基因分型标记集不同且DNA链方向未知。GRAF从使用不同方法获得的基因型数据集中提取10000个信息丰富且独立的单核苷酸多态性(SNP)的基因型,并实现快速算法,使其能够在不到两小时的时间内从dbGaP研究内部和研究之间的88万多个样本中找到所有重复对。此外,GRAF使用两个统计指标,即全基因型错配率(AGMR)和纯合基因型错配率(HGMR),直接从观察到的基因型确定样本关系,而无需估计同源系数(IBD)或亲缘系数的概率,并将预测的关系与系谱文件中报告的关系进行比较。我们用同名的免费C++程序实现了GRAF。在本文中,我们描述了GRAF中的方法,并在来自dbGaP数据库的样本上验证了GRAF的用法。其他科学家可以在他们自己的样本上使用GRAF,并与从dbGaP下载的样本结合使用。