Rusmini Paola, Cristofani Riccardo, Galbiati Mariarita, Cicardi Maria E, Meroni Marco, Ferrari Veronica, Vezzoli Giulia, Tedesco Barbara, Messi Elio, Piccolella Margherita, Carra Serena, Crippa Valeria, Poletti Angelo
Dipartimento di Scienze Farmacologiche e Biomolecolari (DiSFeB), Centro di Eccellenza sulle Malattie Neurodegenerative, Università degli Studi di MilanoMilano, Italy.
Dipartimento di Scienze Biomediche, Metaboliche e Neuroscienze, Università di Modena e Reggio EmiliaModena, Italy.
Front Mol Neurosci. 2017 Jun 21;10:176. doi: 10.3389/fnmol.2017.00176. eCollection 2017.
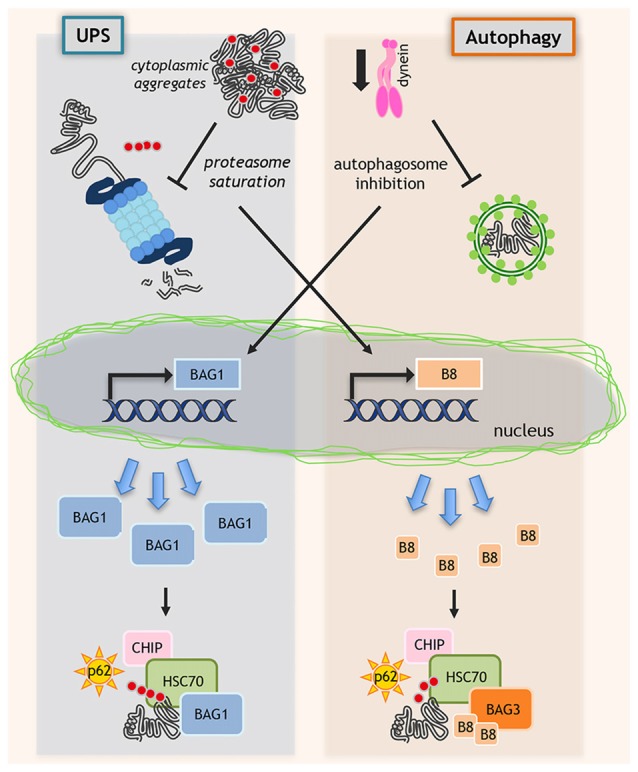
Amyotrophic lateral sclerosis (ALS) and spinal and bulbar muscular atrophy (SBMA) are two motoneuron diseases (MNDs) characterized by aberrant protein behavior in affected cells. In familial ALS (fALS) and in SBMA specific gene mutations lead to the production of neurotoxic proteins or peptides prone to misfold, which then accumulate in form of aggregates. Notably, some of these proteins accumulate into aggregates also in sporadic ALS (sALS) even if not mutated. To prevent proteotoxic stresses detrimental to cells, misfolded and/or aggregated proteins must be rapidly removed by the protein quality control (PQC) system. The small heat shock protein B8 (HSPB8) is a chaperone induced by harmful events, like proteasome inhibition. HSPB8 is expressed both in motoneuron and muscle cells, which are both targets of misfolded protein toxicity in MNDs. In ALS mice models, in presence of the mutant proteins, HSPB8 is upregulated both in spinal cord and muscle. HSPB8 interacts with the HSP70 co-chaperone BAG3 and enhances the degradation of misfolded proteins linked to sALS, or causative of fALS and of SBMA. HSPB8 acts by facilitating autophagy, thereby preventing misfolded protein accumulation in affected cells. BAG3 and BAG1 compete for HSP70-bound clients and target them for disposal to the autophagy or proteasome, respectively. Enhancing the selective targeting of misfolded proteins by HSPB8-BAG3-HSP70 to autophagy may also decrease their delivery to the proteasome by the BAG1-HSP70 complex, thereby limiting possible proteasome overwhelming. Thus, approaches aimed at potentiating HSPB8-BAG3 may contribute to the maintenance of proteostasis and may delay MNDs progression.
肌萎缩侧索硬化症(ALS)和脊髓延髓肌肉萎缩症(SBMA)是两种运动神经元疾病(MNDs),其特征是受影响细胞中存在异常蛋白质行为。在家族性ALS(fALS)和SBMA中,特定基因突变导致产生易于错误折叠的神经毒性蛋白质或肽,然后以聚集体的形式积累。值得注意的是,即使未发生突变,其中一些蛋白质也会在散发性ALS(sALS)中聚集成聚集体。为了防止对细胞有害的蛋白毒性应激,错误折叠和/或聚集的蛋白质必须通过蛋白质质量控制(PQC)系统迅速清除。小热休克蛋白B8(HSPB8)是一种由有害事件(如蛋白酶体抑制)诱导的伴侣蛋白。HSPB8在运动神经元和肌肉细胞中均有表达,而这两种细胞都是MNDs中错误折叠蛋白毒性的靶点。在ALS小鼠模型中,在存在突变蛋白的情况下,HSPB8在脊髓和肌肉中均上调。HSPB8与HSP70共伴侣蛋白BAG3相互作用,并增强与sALS相关的、或导致fALS和SBMA的错误折叠蛋白的降解。HSPB8通过促进自噬发挥作用,从而防止错误折叠蛋白在受影响细胞中积累。BAG3和BAG1竞争与HSP70结合的底物,并分别将它们靶向自噬或蛋白酶体进行处理。增强HSPB8 - BAG3对错误折叠蛋白向自噬的选择性靶向作用,也可能减少它们通过BAG1 - HSP70复合物向蛋白酶体的递送,从而限制可能出现的蛋白酶体过载。因此,旨在增强HSPB8 - BAG3的方法可能有助于维持蛋白质稳态,并可能延缓MNDs的进展。