Henriksen Niel M, Gilson Michael K
Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego , La Jolla, California 92093-0736, United States.
J Chem Theory Comput. 2017 Sep 12;13(9):4253-4269. doi: 10.1021/acs.jctc.7b00359. Epub 2017 Aug 4.
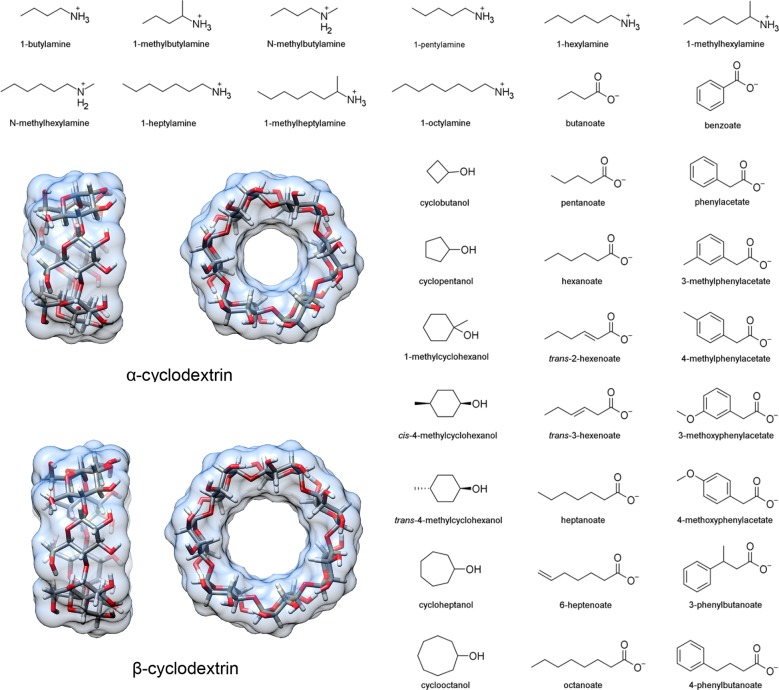
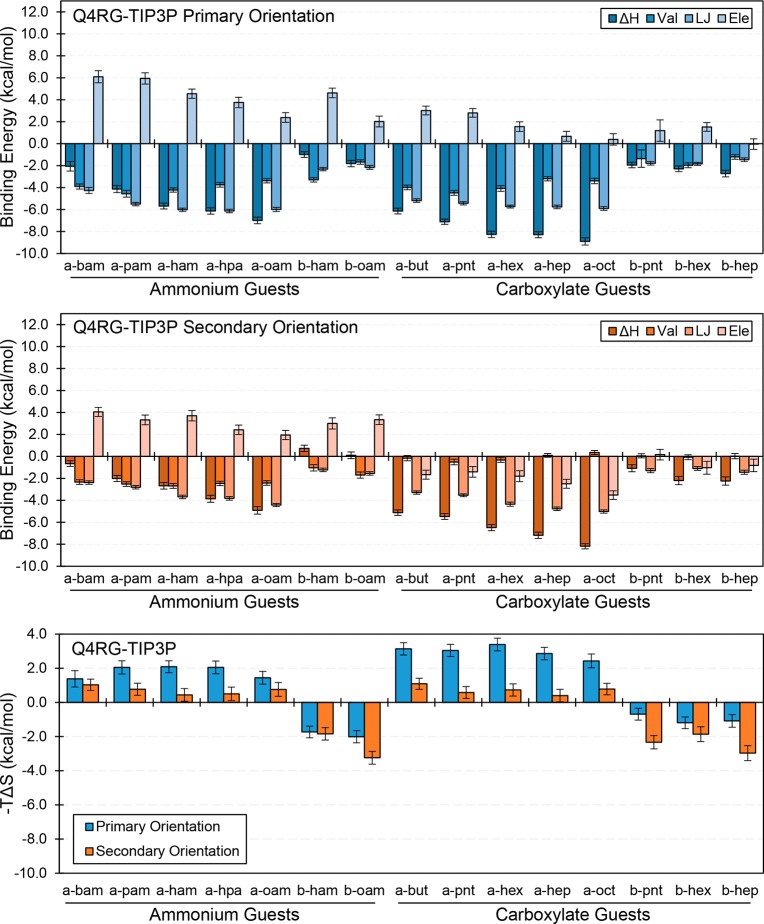
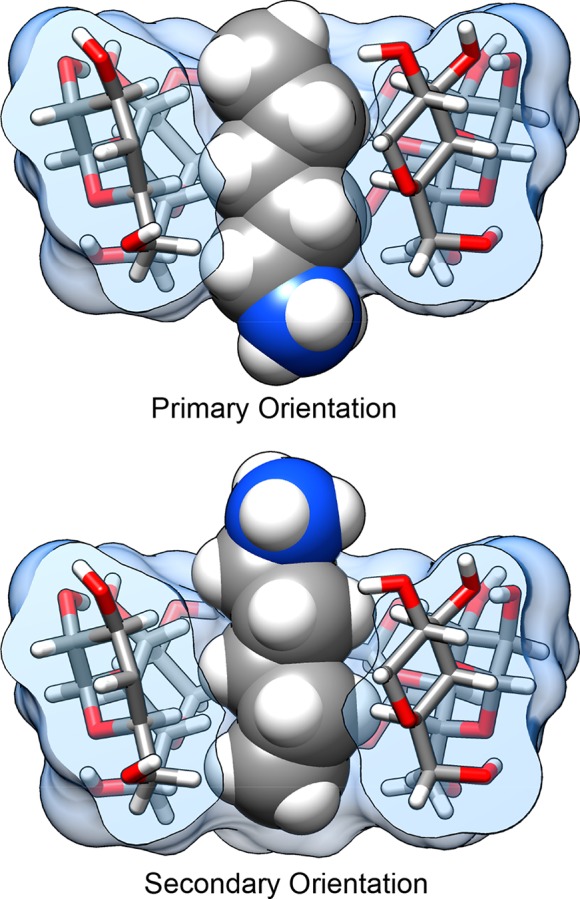
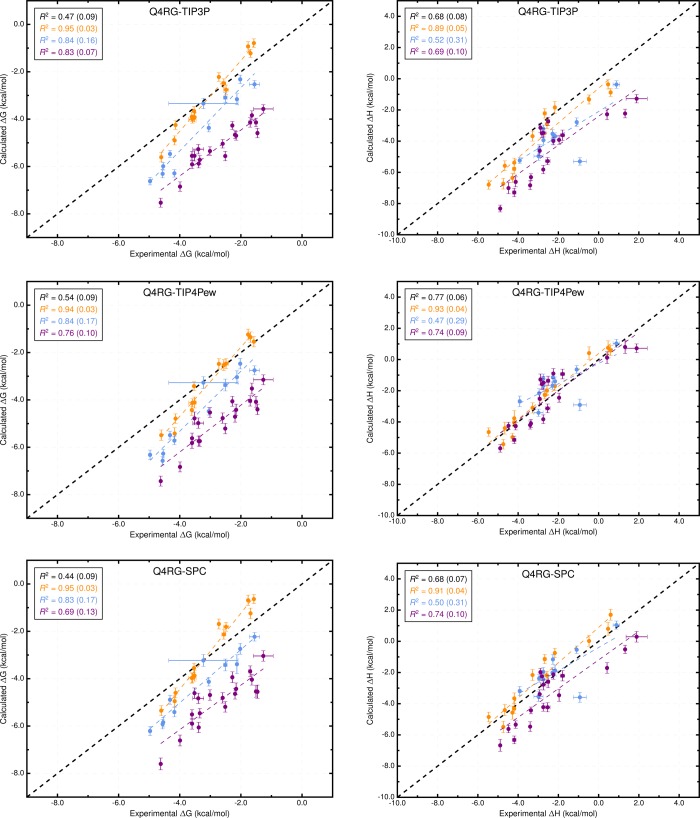
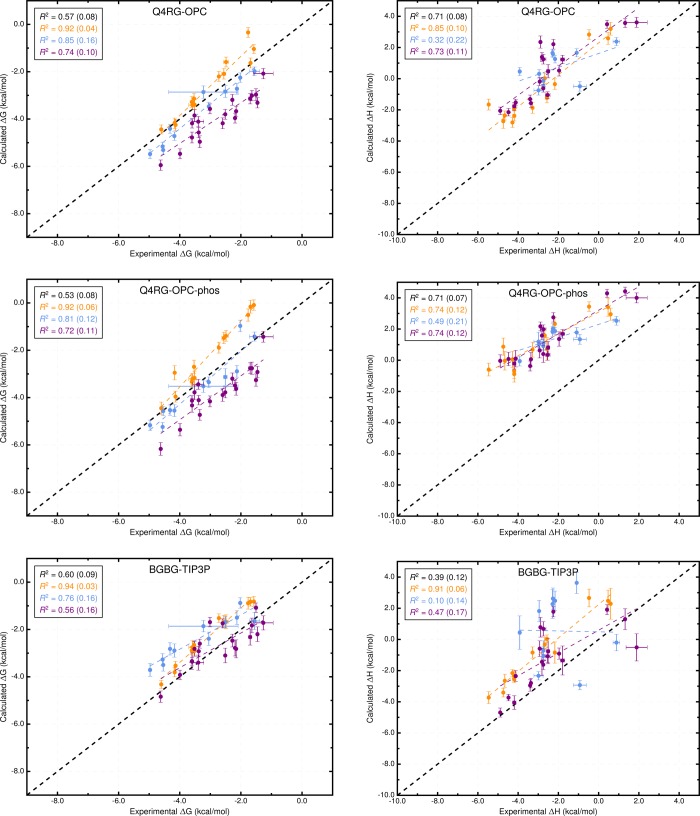
Computational prediction of noncovalent binding free energies with methods based on molecular mechanical force fields has become increasingly routine in drug discovery projects, where they promise to speed the discovery of small molecule ligands to bind targeted proteins with high affinity. Because the reliability of free energy methods still has significant room for improvement, new force fields, or modifications of existing ones, are regularly introduced with the aim of improving the accuracy of molecular simulations. However, comparatively little work has been done to systematically assess how well force fields perform, particularly in relation to the calculation of binding affinities. Hardware advances have made these calculations feasible, but comprehensive force field assessments for protein-ligand sized systems still remain costly. Here, we turn to cyclodextrin host-guest systems, which feature many hallmarks of protein-ligand binding interactions but are generally much more tractable due to their small size. We present absolute binding free energy and enthalpy calculations, using the attach-pull-release (APR) approach, on a set of 43 cyclodextrin-guest pairs for which experimental ITC data are available. The test set comprises both α- and β-cyclodextrin hosts binding a series of small organic guests, each with one of three functional groups: ammonium, alcohol, or carboxylate. Four water models are considered (TIP3P, TIP4Pew, SPC/E, and OPC), along with two partial charge assignment procedures (RESP and AM1-BCC) and two cyclodextrin host force fields. The results suggest a complex set of considerations when choosing a force field for biomolecular simulations. For example, some force field combinations clearly outperform others at the binding enthalpy calculations but not for the binding free energy. Additionally, a force field combination which we expected to be the worst performer gave the most accurate binding free energies - but the least accurate binding enthalpies. The results have implications for the development of improved force fields, and we propose this test set, and potential future elaborations of it, as a powerful validation suite to evaluate new force fields and help guide future force field development.
使用基于分子力学力场的方法对非共价结合自由能进行计算预测,在药物研发项目中已变得越来越常规,在这些项目中,该方法有望加速发现能够以高亲和力结合目标蛋白的小分子配体。由于自由能方法的可靠性仍有很大的提升空间,新的力场或对现有力场的改进经常被引入,目的是提高分子模拟的准确性。然而,在系统评估力场的性能方面,尤其是在结合亲和力的计算方面,所做的工作相对较少。硬件的进步使这些计算变得可行,但对蛋白质 - 配体大小的系统进行全面的力场评估仍然成本高昂。在这里,我们转向环糊精主客体系统,该系统具有蛋白质 - 配体结合相互作用的许多特征,但由于其尺寸小,通常更易于处理。我们使用附着 - 拉动 - 释放(APR)方法,对一组43个有实验ITC数据的环糊精 - 客体对进行了绝对结合自由能和焓的计算。测试集包括α - 和β - 环糊精主体与一系列小有机客体的结合,每个客体具有三种官能团之一:铵基、醇基或羧基。考虑了四种水模型(TIP3P、TIP4Pew、SPC/E和OPC),以及两种部分电荷分配程序(RESP和AM1 - BCC)和两种环糊精主体力场。结果表明,在为生物分子模拟选择力场时需要综合考虑一系列复杂因素。例如,一些力场组合在结合焓的计算中明显优于其他组合,但在结合自由能的计算中并非如此。此外,我们预期表现最差的一个力场组合给出了最准确的结合自由能,但结合焓的准确性最低。这些结果对改进力场的开发具有启示意义,我们提出这个测试集以及未来可能的扩展,作为一个强大的验证套件,用于评估新的力场并帮助指导未来的力场开发。