Liu Jie, Su Minyi, Liu Zhihai, Li Jie, Li Yan, Wang Renxiao
State Key Laboratory of Bioorganic and Natural Products Chemistry, Collaborative Innovation Center of Chemistry for Life Sciences, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai, 200032, China.
State Key Laboratory of Quality Research in Chinese Medicine, Macau Institute for Applied Research in Medicine and Health, Macau University of Science and Technology, Macau, People's Republic of China.
BMC Bioinformatics. 2017 Jul 18;18(1):343. doi: 10.1186/s12859-017-1750-5.
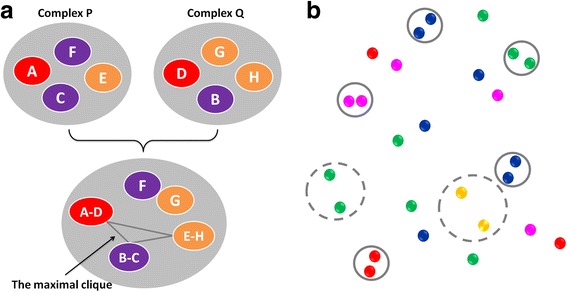
In structure-based drug design, binding affinity prediction remains as a challenging goal for current scoring functions. Development of target-biased scoring functions provides a new possibility for tackling this problem, but this approach is also associated with certain technical difficulties. We previously reported the Knowledge-Guided Scoring (KGS) method as an alternative approach (BMC Bioinformatics, 2010, 11, 193-208). The key idea is to compute the binding affinity of a given protein-ligand complex based on the known binding data of an appropriate reference complex, so the error in binding affinity prediction can be reduced effectively.
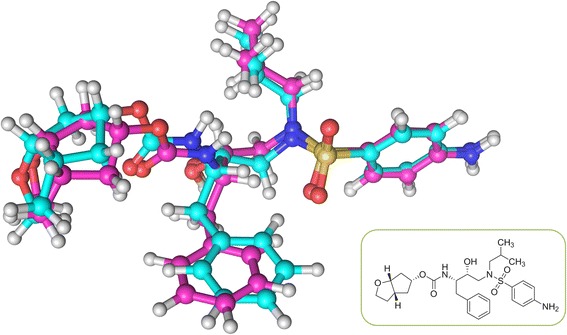
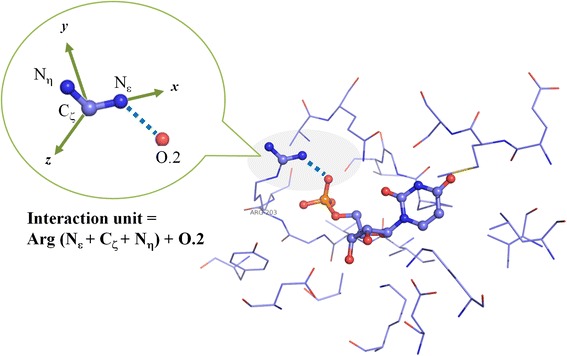
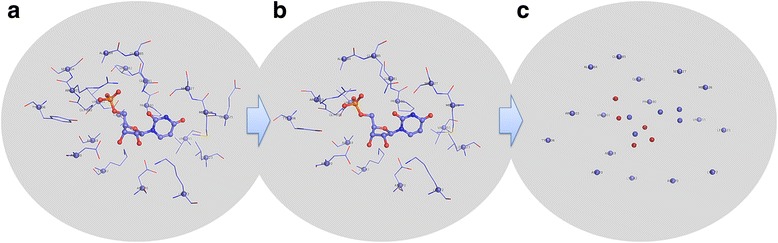
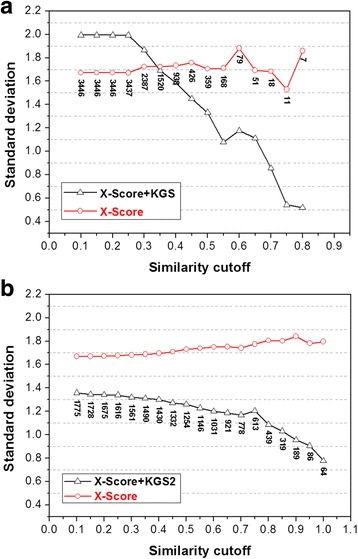
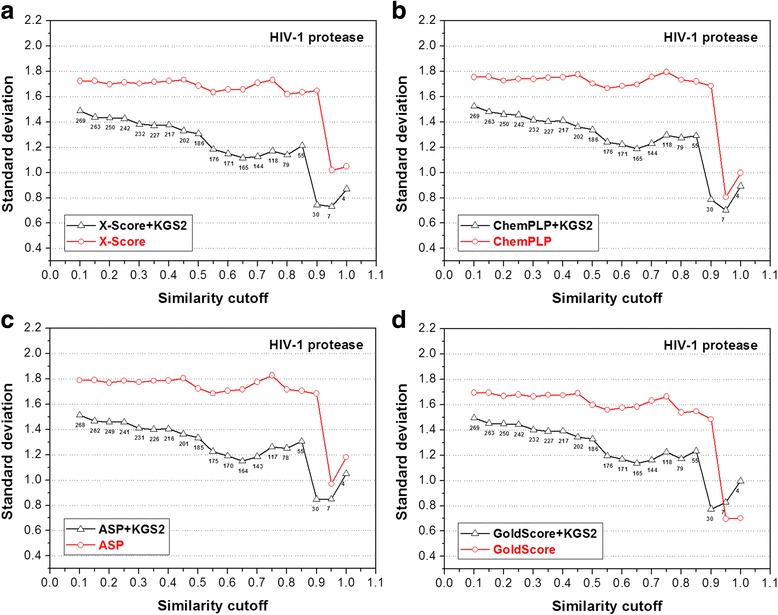
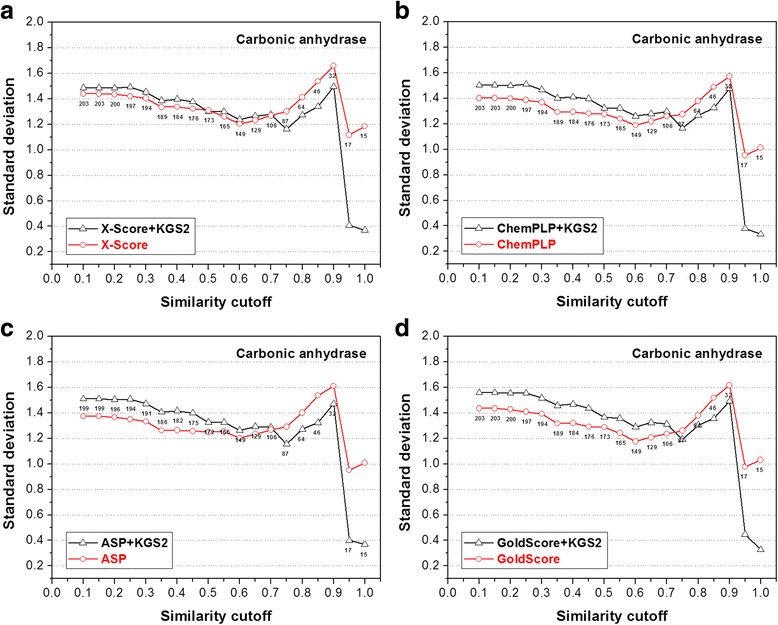
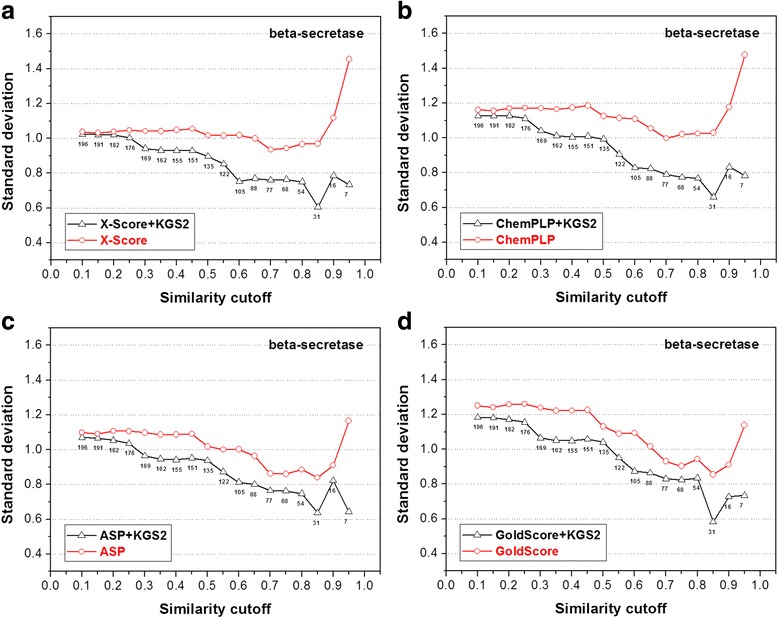
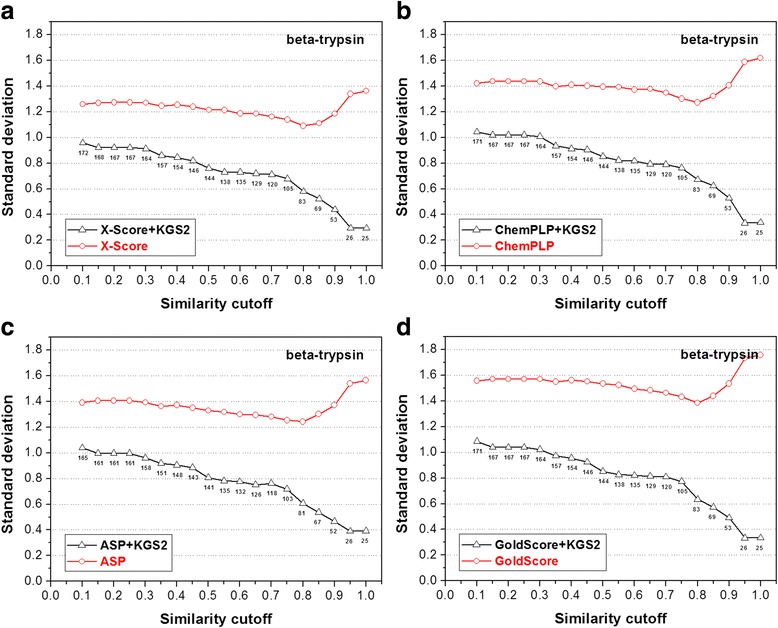
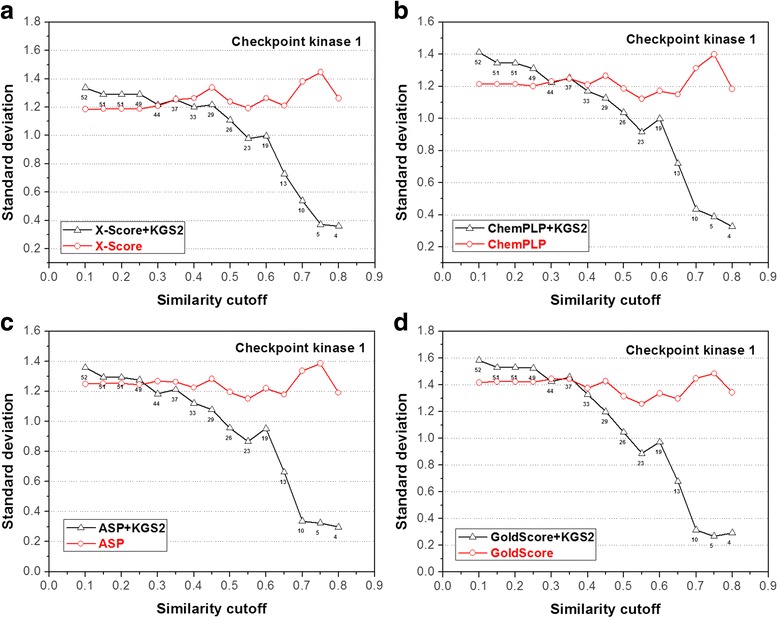
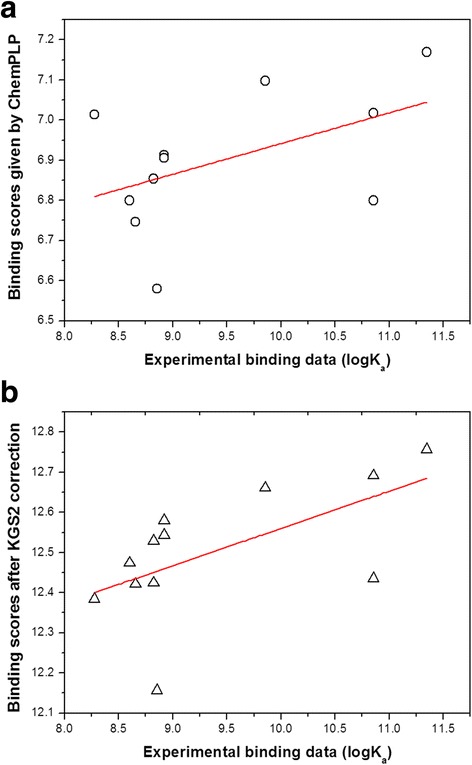
In this study, we have developed an upgraded version, i.e. KGS2, by employing 3D protein-ligand interaction fingerprints in reference selection. KGS2 was evaluated in combination with four scoring functions (X-Score, ChemPLP, ASP, and GoldScore) on five drug targets (HIV-1 protease, carbonic anhydrase 2, beta-secretase 1, beta-trypsin, and checkpoint kinase 1). In the in situ scoring test, considerable improvements were observed in most cases after application of KGS2. Besides, the performance of KGS2 was always better than KGS in all cases. In the more challenging molecular docking test, application of KGS2 also led to improved structure-activity relationship in some cases.
KGS2 can be applied as a convenient "add-on" to current scoring functions without the need to re-engineer them, and its application is not limited to certain target proteins as customized scoring functions. As an interpolation method, its accuracy in principle can be improved further with the increasing knowledge of protein-ligand complex structures and binding affinity data. We expect that KGS2 will become a practical tool for enhancing the performance of current scoring functions in binding affinity prediction. The KGS2 software is available upon contacting the authors.
在基于结构的药物设计中,结合亲和力预测对于当前的评分函数而言仍是一个具有挑战性的目标。开发针对靶点的评分函数为解决这一问题提供了新的可能性,但这种方法也存在一定的技术难题。我们之前报道了知识引导评分(KGS)方法作为一种替代方法(《BMC生物信息学》,2010年,11卷,193 - 208页)。其关键思想是基于合适参考复合物的已知结合数据来计算给定蛋白质 - 配体复合物的结合亲和力,从而有效降低结合亲和力预测中的误差。
在本研究中,我们通过在参考选择中采用三维蛋白质 - 配体相互作用指纹图谱开发了一个升级版,即KGS2。KGS2与四种评分函数(X - Score、ChemPLP、ASP和GoldScore)相结合,在五个药物靶点(HIV - 1蛋白酶、碳酸酐酶2、β - 分泌酶1、β - 胰蛋白酶和检查点激酶1)上进行了评估。在原位评分测试中,应用KGS2后,多数情况下观察到了显著改善。此外,在所有情况下,KGS2的性能始终优于KGS。在更具挑战性的分子对接测试中,应用KGS2在某些情况下也改善了构效关系。
KGS2可以作为一种便捷的“附加”方法应用于当前的评分函数,无需对其进行重新设计,并且其应用不像定制评分函数那样局限于特定的靶蛋白。作为一种插值方法,原则上随着对蛋白质 - 配体复合物结构和结合亲和力数据的了解不断增加,其准确性可以进一步提高。我们期望KGS2将成为提高当前评分函数在结合亲和力预测中性能的实用工具。可通过联系作者获取KGS2软件。