Grofe Adam, Chen Xin, Liu Wenjian, Gao Jiali
Theoretical Chemistry Institute, Jilin University , Changchun, Jilin Province 130023, People's Republic of China.
Department of Chemistry, University of Minnesota , Minneapolis, Minnesota 55455, United States.
J Phys Chem Lett. 2017 Oct 5;8(19):4838-4845. doi: 10.1021/acs.jpclett.7b02202. Epub 2017 Sep 22.
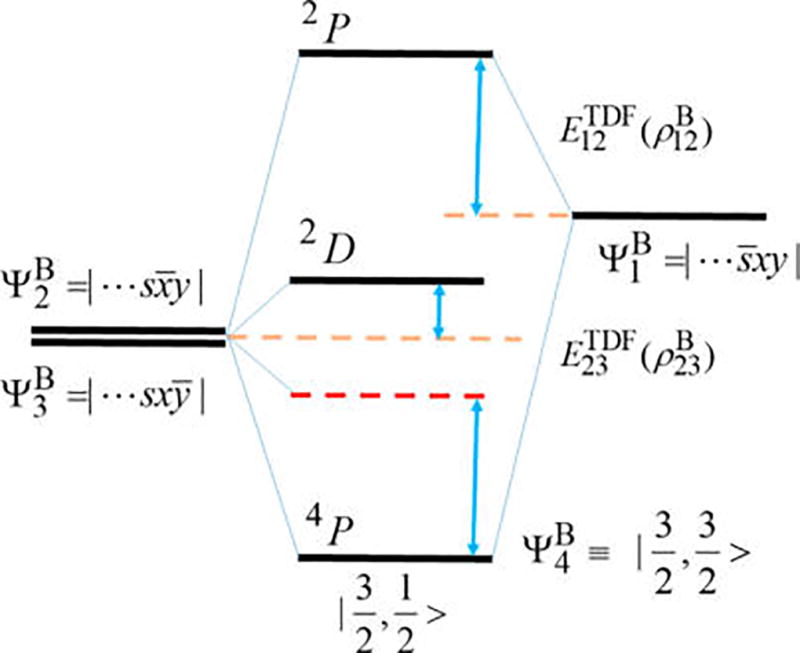
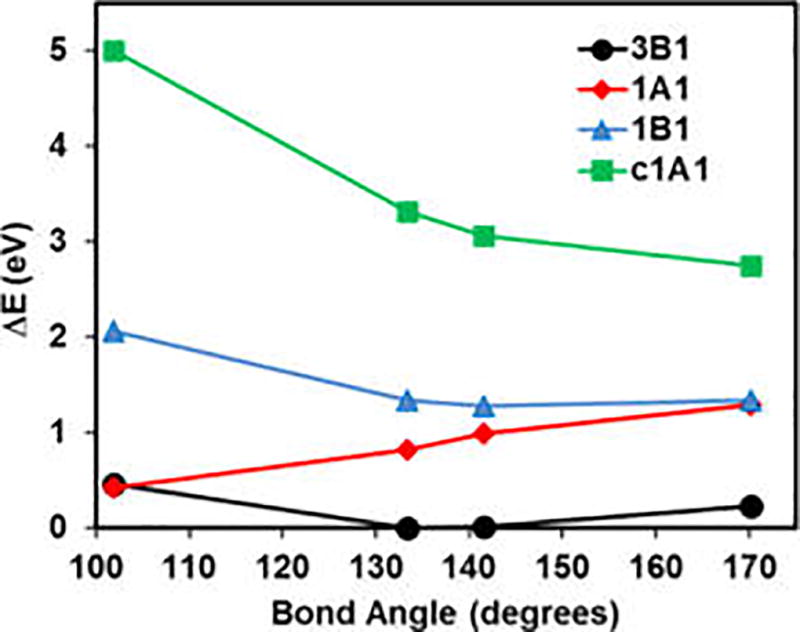
Kohn-Sham density functional theory has been tremendously successful in chemistry and physics. Yet, it is unable to describe the energy degeneracy of spin-multiplet components with any approximate functional. This work features two contributions. (1) We present a multistate density functional theory (MSDFT) to represent spin-multiplet components and to determine multiplet energies. MSDFT is a hybrid approach, taking advantage of both wave function theory and density functional theory. Thus, the wave functions, electron densities and energy density-functionals for ground and excited states and for different components are treated on the same footing. The method is illustrated on valence excitations of atoms and molecules. (2) Importantly, a key result is that for cases in which the high-spin components can be determined separately by Kohn-Sham density functional theory, the transition density functional in MSDFT (which describes electronic coupling) can be defined rigorously. The numerical results may be explored to design and optimize transition density functionals for configuration coupling in multiconfigurational DFT.
科恩-沈(Kohn-Sham)密度泛函理论在化学和物理学领域取得了巨大成功。然而,它无法用任何近似泛函描述自旋多重态组分的能量简并性。这项工作有两个贡献。(1)我们提出了一种多态密度泛函理论(MSDFT)来表示自旋多重态组分并确定多重态能量。MSDFT是一种混合方法,利用了波函数理论和密度泛函理论。因此,基态和激发态以及不同组分的波函数、电子密度和能量密度泛函都在相同的基础上进行处理。该方法通过原子和分子的价激发进行了说明。(2)重要的是,一个关键结果是,对于高自旋组分可由科恩-沈密度泛函理论单独确定的情况,MSDFT中的跃迁密度泛函(它描述电子耦合)可以被严格定义。数值结果可用于探索设计和优化多组态密度泛函理论中用于组态耦合的跃迁密度泛函。