Zytnicki Matthias
MIAT, Toulouse INRA, BP 52627, Castanet-Tolosan cedex, 31326, France.
BMC Bioinformatics. 2017 Sep 15;18(1):411. doi: 10.1186/s12859-017-1816-4.
RNA-Seq is currently used routinely, and it provides accurate information on gene transcription. However, the method cannot accurately estimate duplicated genes expression. Several strategies have been previously used (drop duplicated genes, distribute uniformly the reads, or estimate expression), but all of them provide biased results.
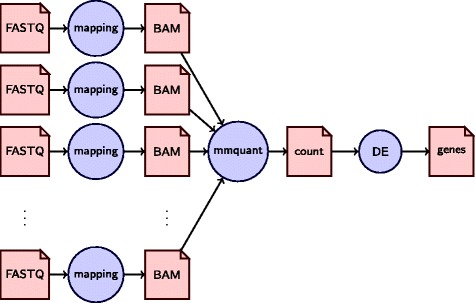
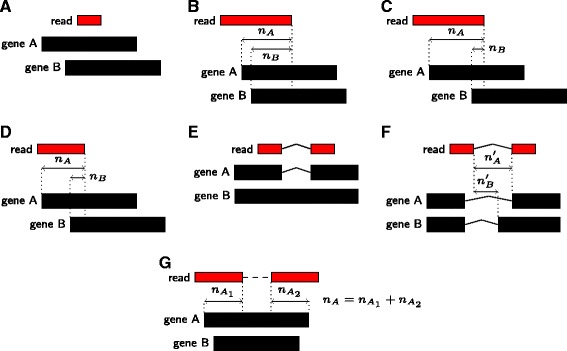
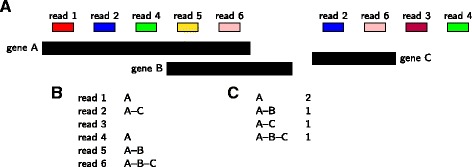
We provide here a tool, called mmquant, for computing gene expression, included duplicated genes. If a read maps at different positions, the tool detects that the corresponding genes are duplicated; it merges the genes and creates a merged gene. The counts of ambiguous reads is then based on the input genes and the merged genes.
mmquant is a drop-in replacement of the widely used tools htseq-count and featureCounts that handles multi-mapping reads in an unabiased way.
RNA测序目前已被常规使用,它能提供有关基因转录的准确信息。然而,该方法无法准确估计重复基因的表达。此前已经使用了几种策略(去除重复基因、均匀分配 reads 或估计表达量),但所有这些策略都会产生有偏差的结果。
我们在此提供了一种名为mmquant的工具,用于计算基因表达量,包括重复基因。如果一条 reads 映射到不同位置,该工具会检测到相应的基因是重复的;它会合并这些基因并创建一个合并基因。然后,模糊 reads 的计数基于输入基因和合并基因。
mmquant是广泛使用的工具htseq-count和featureCounts的直接替代品,它能以无偏差的方式处理多映射 reads。