Genomic Research Laboratory, Service of Infectious Diseases, Geneva University Hospitals (HUG), 1205 Geneva, Switzerland.
Service of Infectious Diseases, Geneva University Hospitals and Faculty of Medicine, 1205 Geneva, Switzerland.
Int J Mol Sci. 2017 Sep 20;18(9):2011. doi: 10.3390/ijms18092011.
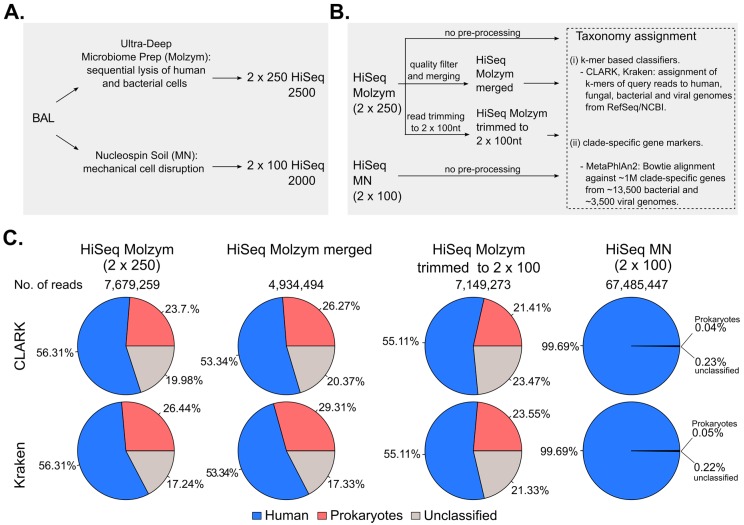
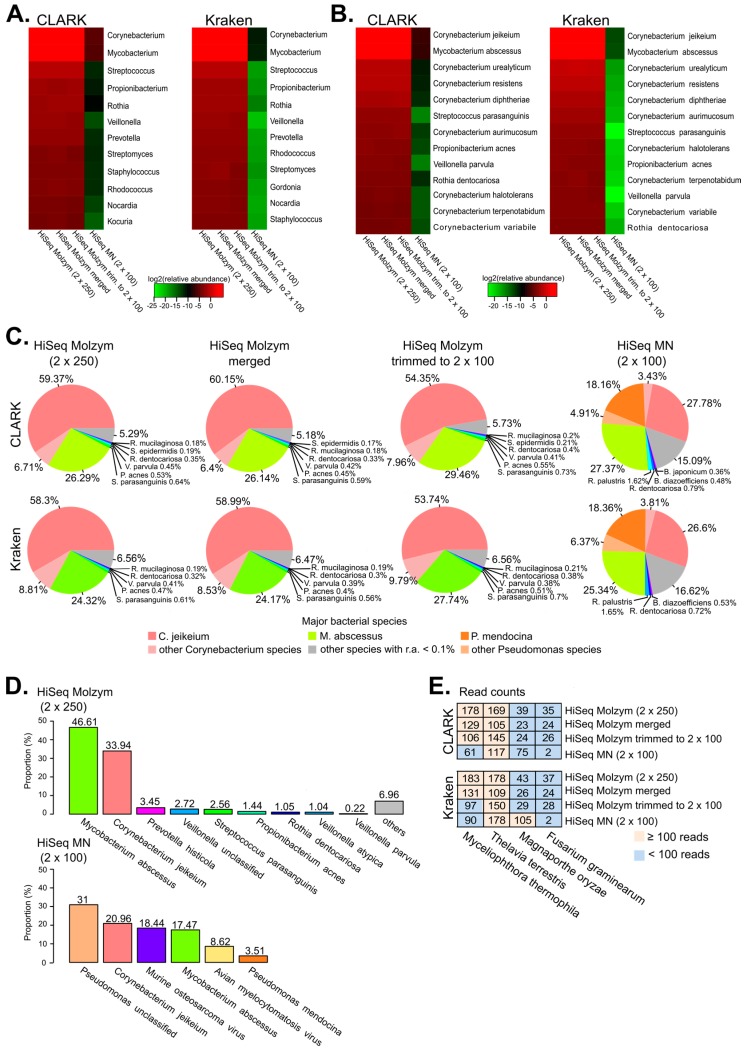
The applications of whole-metagenome shotgun sequencing (WMGS) in routine clinical analysis are still limited. A combination of a DNA extraction procedure, sequencing, and bioinformatics tools is essential for the removal of human DNA and for improving bacterial species identification in a timely manner. We tackled these issues with a broncho-alveolar lavage (BAL) sample from an immunocompromised patient who had developed severe chronic pneumonia. We extracted DNA from the BAL sample with protocols based either on sequential lysis of human and bacterial cells or on the mechanical disruption of all cells. Metagenomic libraries were sequenced on Illumina HiSeq platforms. Microbial community composition was determined by k-mer analysis or by mapping to taxonomic markers. Results were compared to those obtained by conventional clinical culture and molecular methods. Compared to mechanical cell disruption, a sequential lysis protocol resulted in a significantly increased proportion of bacterial DNA over human DNA and higher sequence coverage of , and , the bacteria reported by clinical microbiology tests. In addition, we identified anaerobic bacteria not searched for by the clinical laboratory. Our results further support the implementation of WMGS in clinical routine diagnosis for bacterial identification.
全基因组鸟枪法测序(WMGS)在常规临床分析中的应用仍然有限。对于去除人 DNA 和及时提高细菌物种鉴定的准确性,需要结合 DNA 提取程序、测序和生物信息学工具。我们使用免疫功能低下患者的支气管肺泡灌洗液(BAL)样本解决了这些问题,该患者患有严重的慢性肺炎。我们使用基于依次裂解人和细菌细胞或机械破坏所有细胞的方案从 BAL 样本中提取 DNA。将 metagenomic 文库在 Illumina HiSeq 平台上进行测序。通过 k-mer 分析或映射到分类标记物来确定微生物群落组成。将结果与传统的临床培养和分子方法的结果进行比较。与机械细胞破坏相比,顺序裂解方案导致细菌 DNA 相对于人 DNA 的比例显著增加,并且细菌的序列覆盖率更高, 和 ,这是临床微生物学检测报告的细菌。此外,我们还鉴定了临床实验室未搜索到的厌氧菌。我们的结果进一步支持在临床常规诊断中实施 WMGS 进行细菌鉴定。