Human Longevity Inc., San Diego, CA, 92121, USA.
J. Craig Venter Institute, La Jolla, CA, 92037, USA.
Sci Rep. 2018 Mar 12;8(1):4333. doi: 10.1038/s41598-018-22660-8.
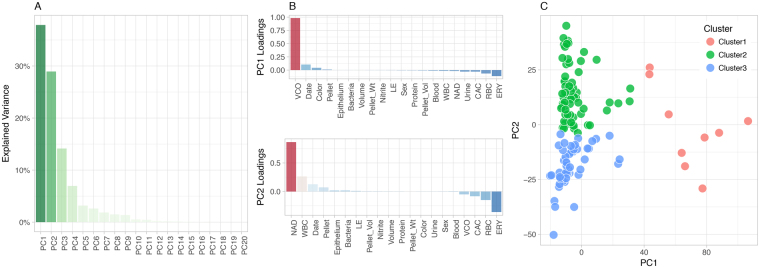
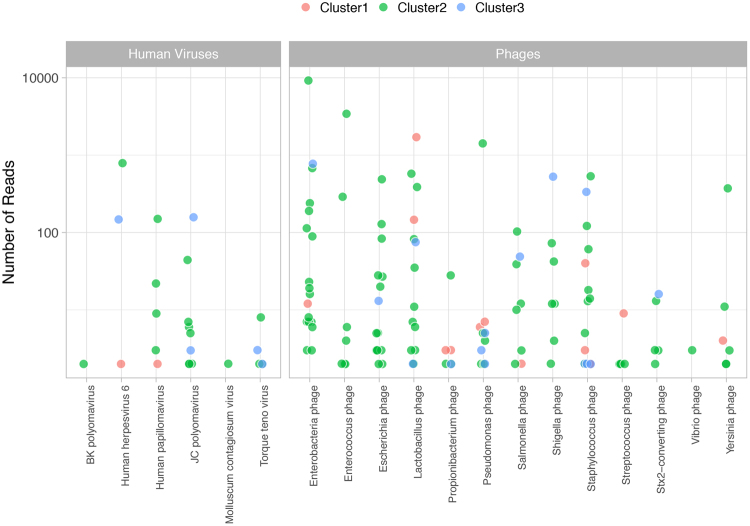
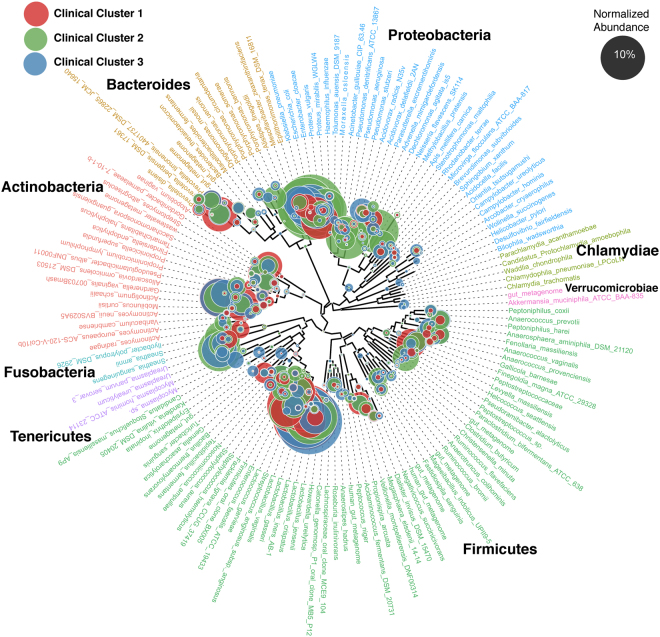
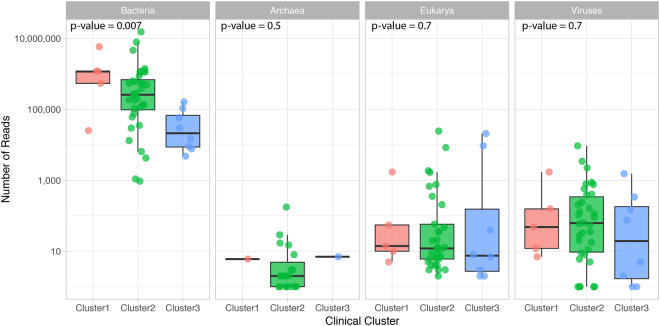
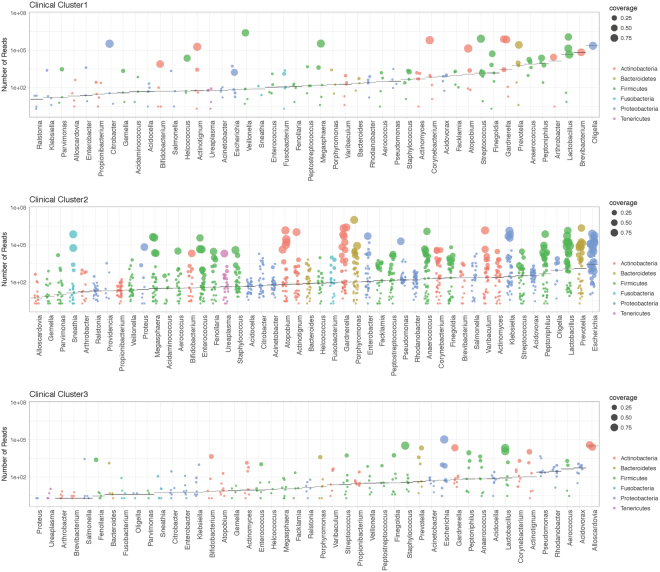
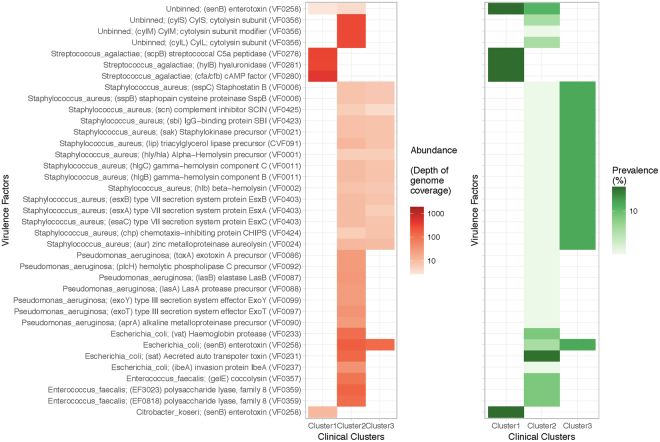
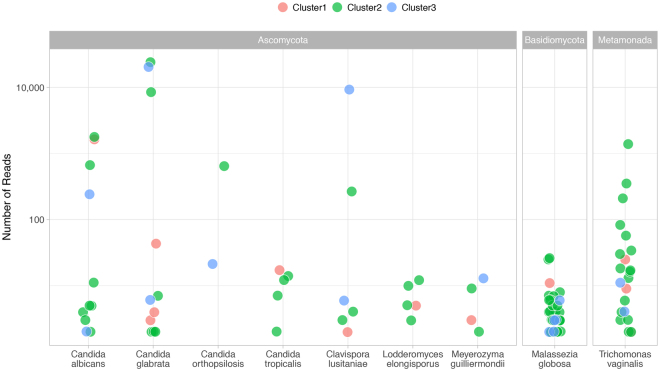
Urine culture and microscopy techniques are used to profile the bacterial species present in urinary tract infections. To gain insight into the urinary flora, we analyzed clinical laboratory features and the microbial metagenome of 121 clean-catch urine samples. 16S rDNA gene signatures were successfully obtained for 116 participants, while metagenome sequencing data was successfully generated for samples from 49 participants. Although 16S rDNA sequencing was more sensitive, metagenome sequencing allowed for a more comprehensive and unbiased representation of the microbial flora, including eukarya and viral pathogens, and of bacterial virulence factors. Urine samples positive by metagenome sequencing contained a plethora of bacterial (median 41 genera/sample), eukarya (median 2 species/sample) and viral sequences (median 3 viruses/sample). Genomic analyses suggested cases of infection with potential pathogens that are often missed during routine urine culture due to species specific growth requirements. While conventional microbiological methods are inadequate to identify a large diversity of microbial species that are present in urine, genomic approaches appear to more comprehensively and quantitatively describe the urinary microbiome.
尿培养和显微镜技术用于分析尿路感染中存在的细菌种类。为了深入了解尿中的菌群,我们分析了 121 例清洁尿样的临床实验室特征和微生物宏基因组。成功获得了 116 名参与者的 16S rDNA 基因特征,而 49 名参与者的样本成功生成了宏基因组测序数据。尽管 16S rDNA 测序更敏感,但宏基因组测序可以更全面、更公正地代表微生物菌群,包括真核生物和病毒病原体,以及细菌毒力因子。通过宏基因组测序呈阳性的尿液样本包含大量细菌(中位数为每个样本 41 个属)、真核生物(中位数为每个样本 2 个种)和病毒序列(中位数为每个样本 3 种病毒)。基因组分析提示存在感染潜在病原体的情况,而这些病原体由于特定的生长要求,在常规尿培养中经常被遗漏。虽然传统的微生物学方法不足以识别尿液中存在的大量微生物种类,但基因组方法似乎更全面和定量地描述了尿微生物组。