Department of Botany and Plant Pathology and Center for Genome Research and Biocomputing, Oregon State University, Corvallis, OR 97331, United States.
National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894, United States.
Virus Res. 2018 Jan 15;244:36-52. doi: 10.1016/j.virusres.2017.10.020. Epub 2017 Nov 8.
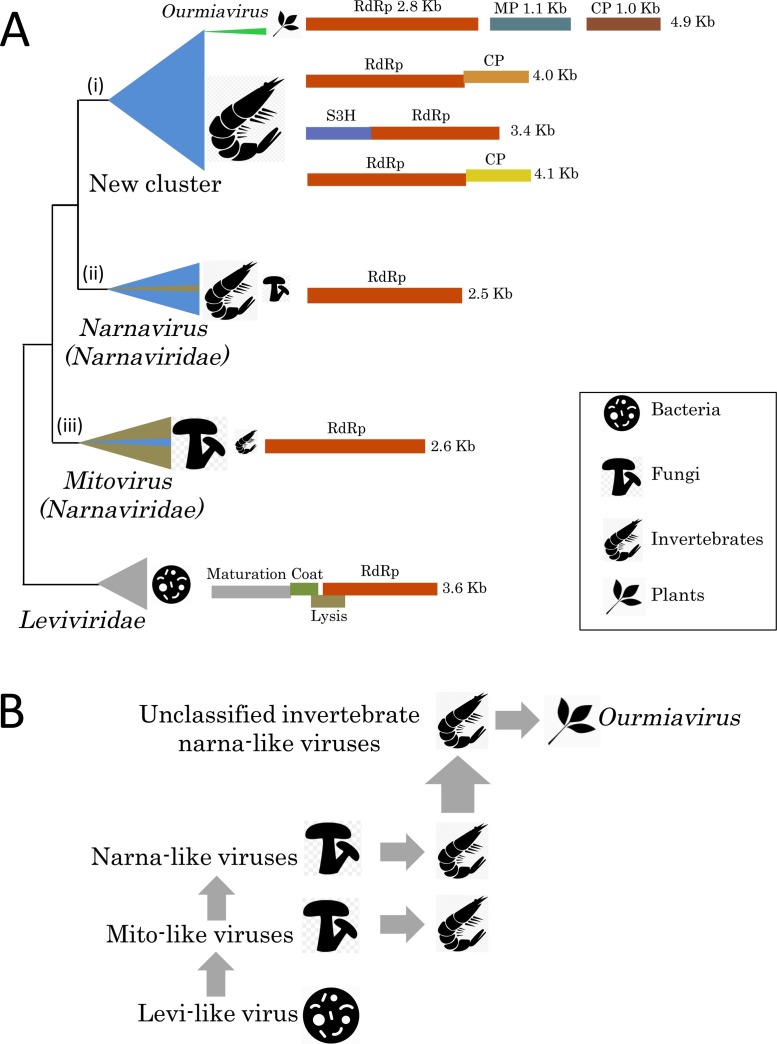
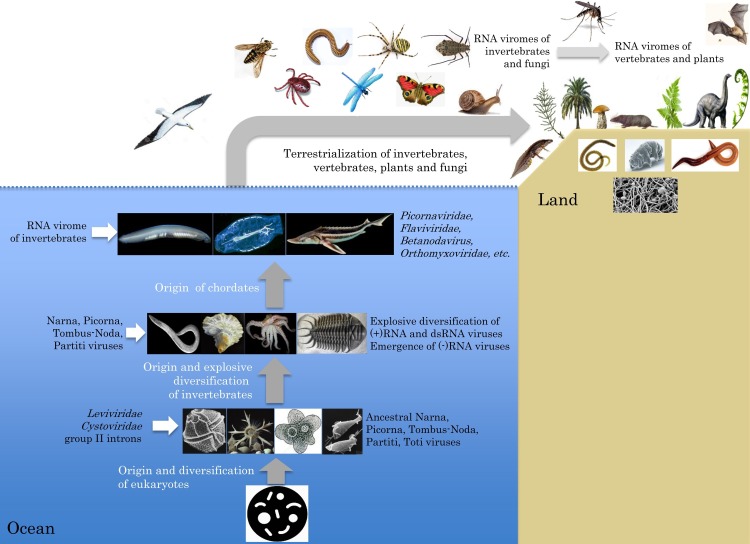
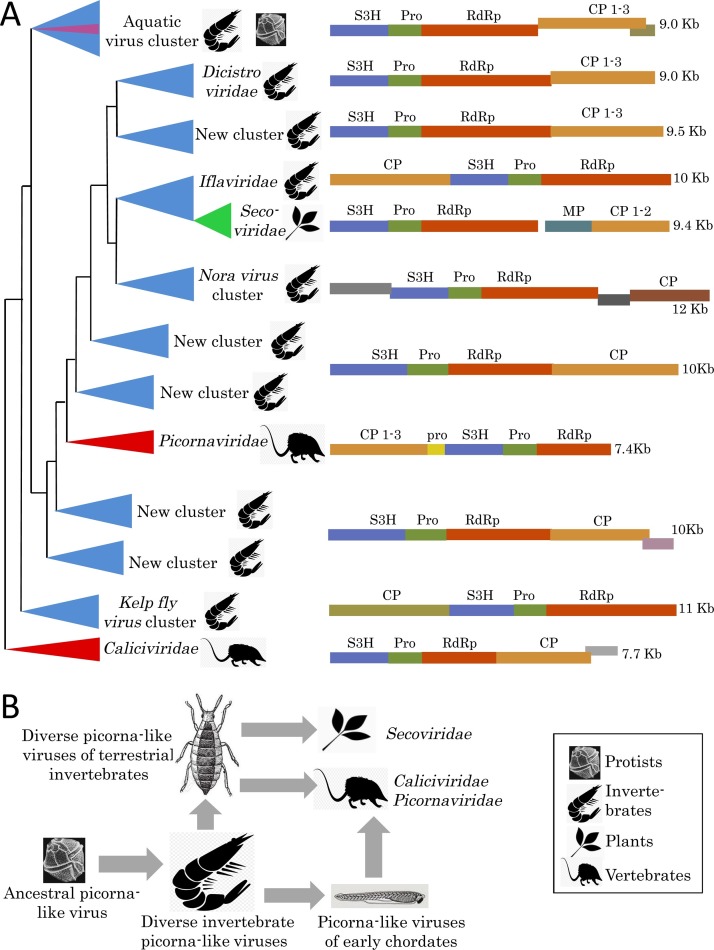
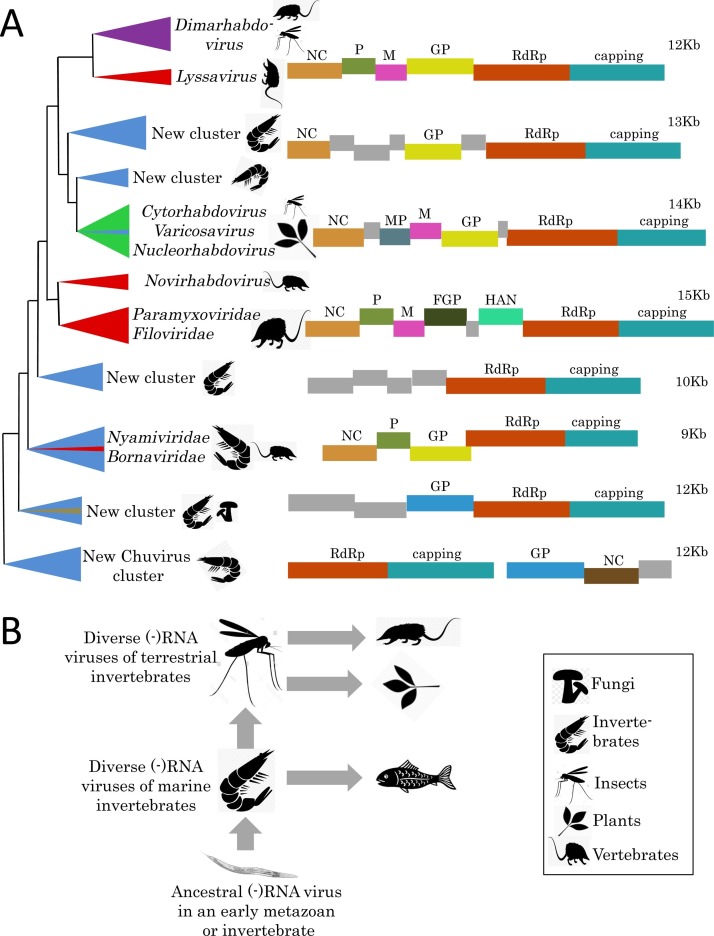
Virus metagenomics is a young research filed but it has already transformed our understanding of virus diversity and evolution, and illuminated at a new level the connections between virus evolution and the evolution and ecology of the hosts. In this review article, we examine the new picture of the evolution of RNA viruses, the dominant component of the eukaryotic virome, that is emerging from metagenomic data analysis. The major expansion of many groups of RNA viruses through metagenomics allowed the construction of substantially improved phylogenetic trees for the conserved virus genes, primarily, the RNA-dependent RNA polymerases (RdRp). In particular, a new superfamily of widespread, small positive-strand RNA viruses was delineated that unites tombus-like and noda-like viruses. Comparison of the genome architectures of RNA viruses discovered by metagenomics and by traditional methods reveals an extent of gene module shuffling among diverse virus genomes that far exceeds the previous appreciation of this evolutionary phenomenon. Most dramatically, inclusion of the metagenomic data in phylogenetic analyses of the RdRp resulted in the identification of numerous, strongly supported groups that encompass RNA viruses from diverse hosts including different groups of protists, animals and plants. Notwithstanding potential caveats, in particular, incomplete and uneven sampling of eukaryotic taxa, these highly unexpected findings reveal horizontal virus transfer (HVT) between diverse hosts as the central aspect of RNA virus evolution. The vast and diverse virome of invertebrates, particularly nematodes and arthropods, appears to be the reservoir, from which the viromes of plants and vertebrates evolved via multiple HVT events.
病毒宏基因组学是一个年轻的研究领域,但它已经改变了我们对病毒多样性和进化的理解,并在新的层面上阐明了病毒进化与宿主进化和生态学之间的联系。在这篇综述文章中,我们考察了从宏基因组数据分析中浮现出的真核病毒组中占主导地位的 RNA 病毒进化的新图景。通过宏基因组学,许多 RNA 病毒群的大规模扩张使得对保守病毒基因(主要是 RNA 依赖性 RNA 聚合酶(RdRp))的系统发育树的构建得到了极大的改进。特别是,定义了一个广泛存在的、小型正链 RNA 病毒的新超家族,将 T4-like 和 noda-like 病毒联合在一起。通过宏基因组学和传统方法发现的 RNA 病毒的基因组结构比较揭示了不同病毒基因组之间基因模块重组的程度远远超过了对这种进化现象的先前认识。最引人注目的是,将宏基因组数据纳入 RdRp 的系统发育分析中,鉴定出了许多强烈支持的群体,其中包括来自不同宿主的 RNA 病毒,包括不同组的原生动物、动物和植物。尽管存在潜在的警告,特别是真核生物分类群的不完全和不均匀采样,但这些出人意料的发现揭示了水平病毒转移(HVT)是 RNA 病毒进化的核心方面。无脊椎动物(特别是线虫和节肢动物)的庞大而多样的病毒组似乎是植物和脊椎动物病毒组通过多次 HVT 事件进化而来的储库。