Sloma Michael F, Mathews David H
Department of Biochemistry & Biophysics and Center for RNA Biology, University of Rochester Medical Center, Rochester, NY, United States of America.
Department of Biostatistics & Computational Biology, University of Rochester Medical Center, Rochester, NY, United States of America.
PLoS Comput Biol. 2017 Nov 6;13(11):e1005827. doi: 10.1371/journal.pcbi.1005827. eCollection 2017 Nov.
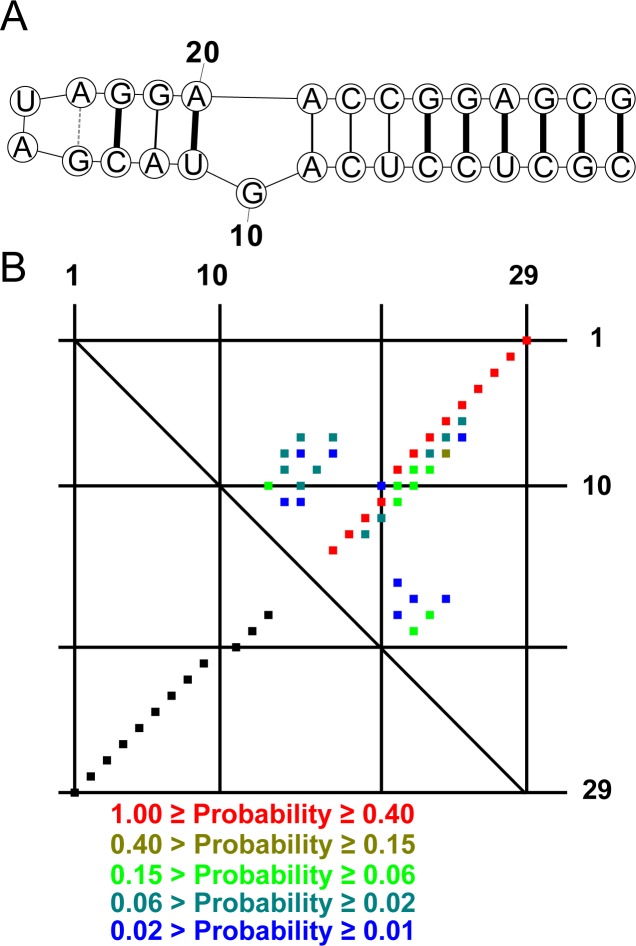
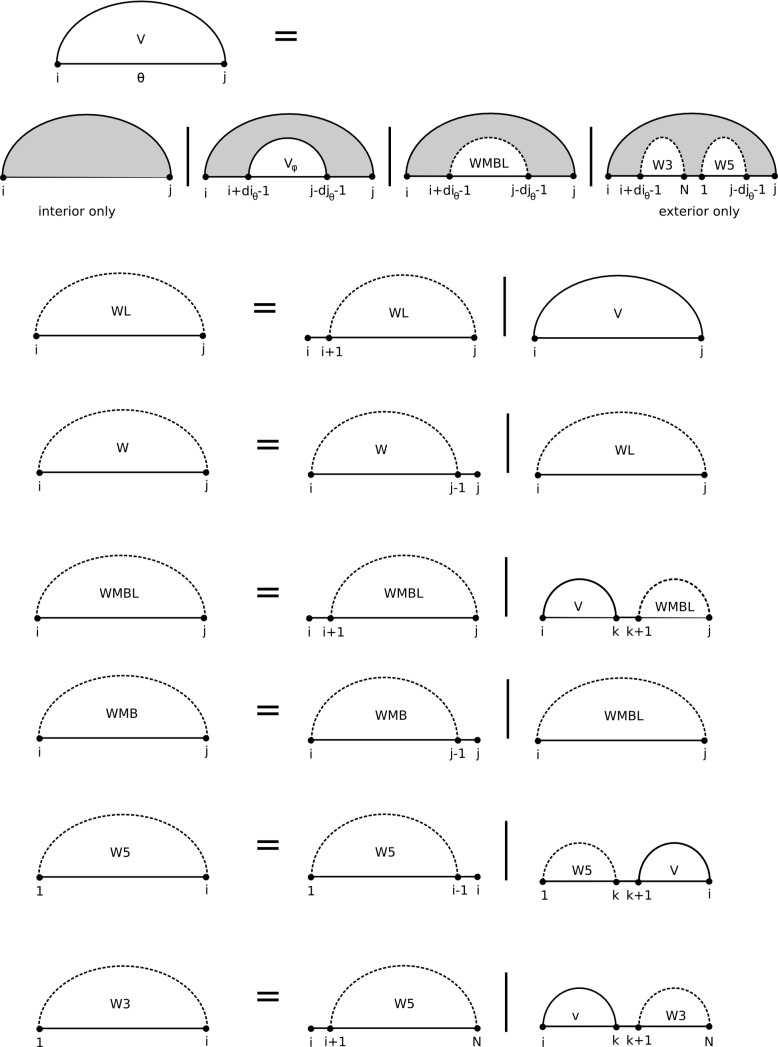
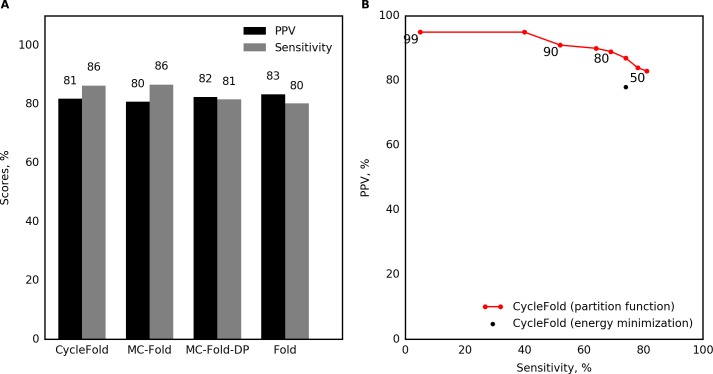
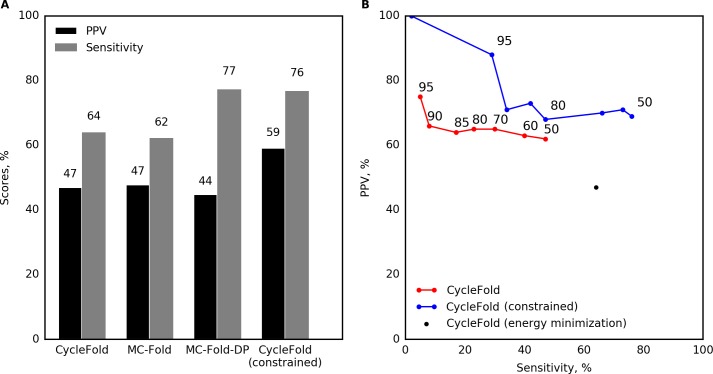
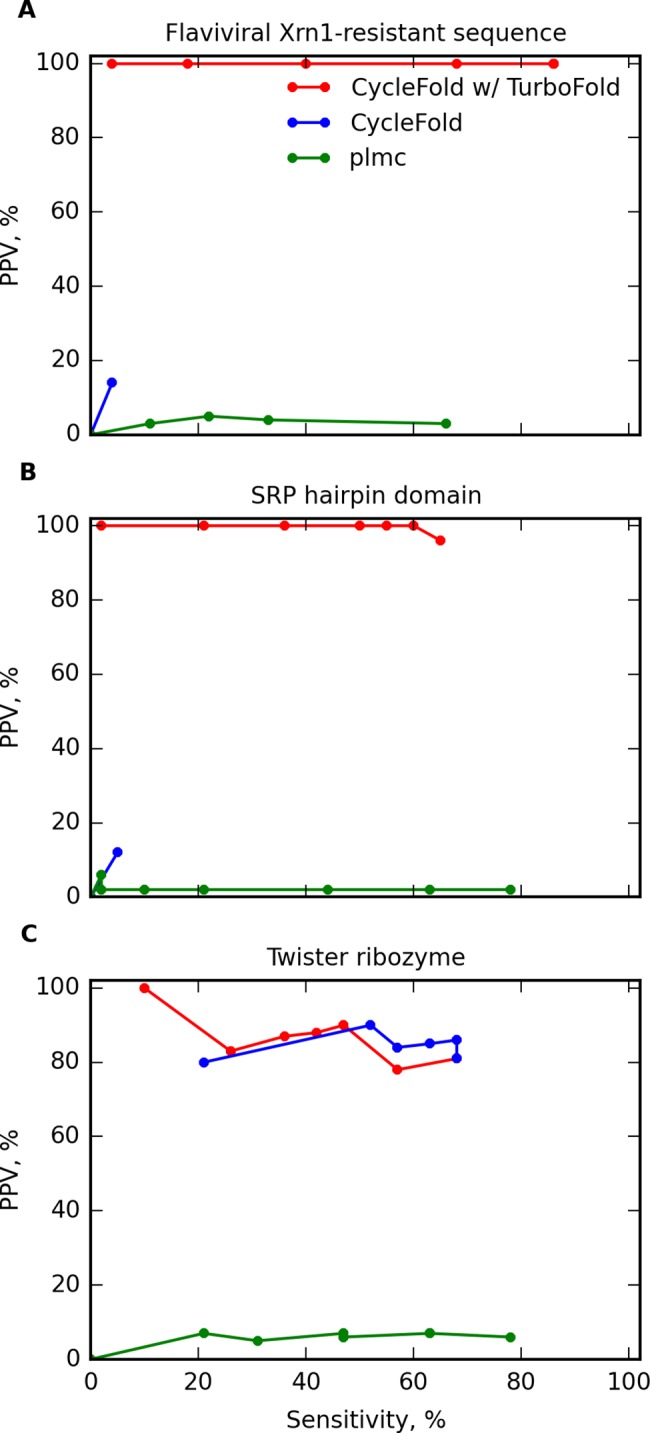
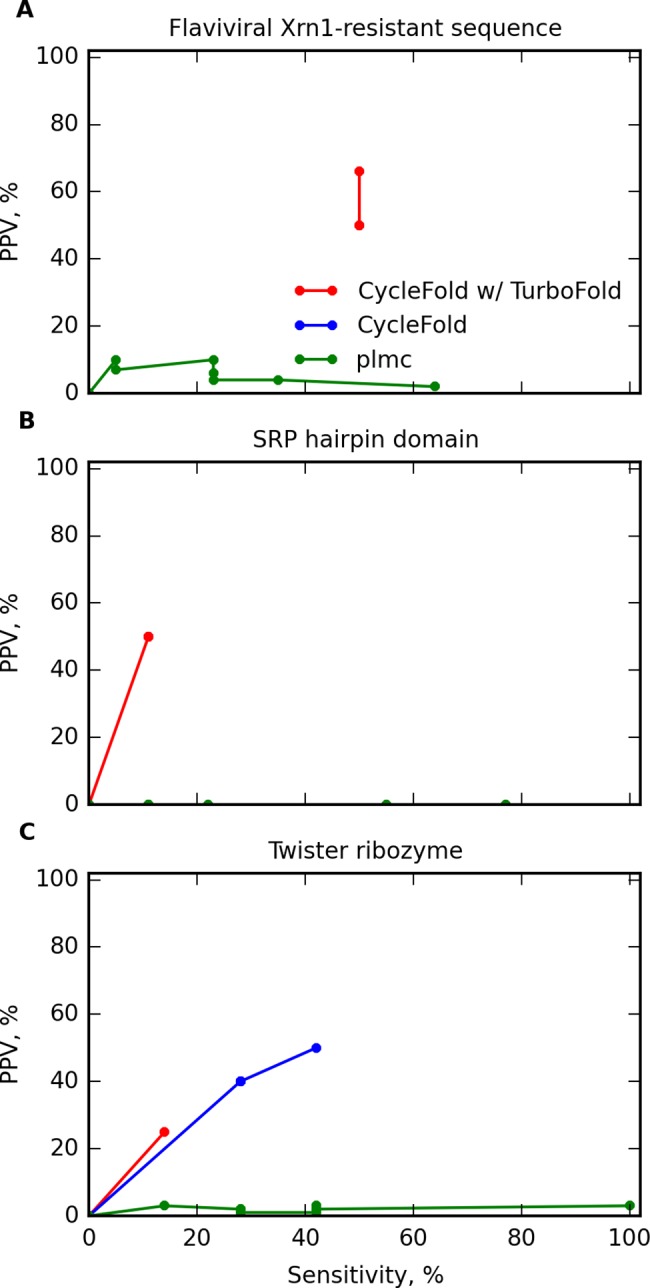
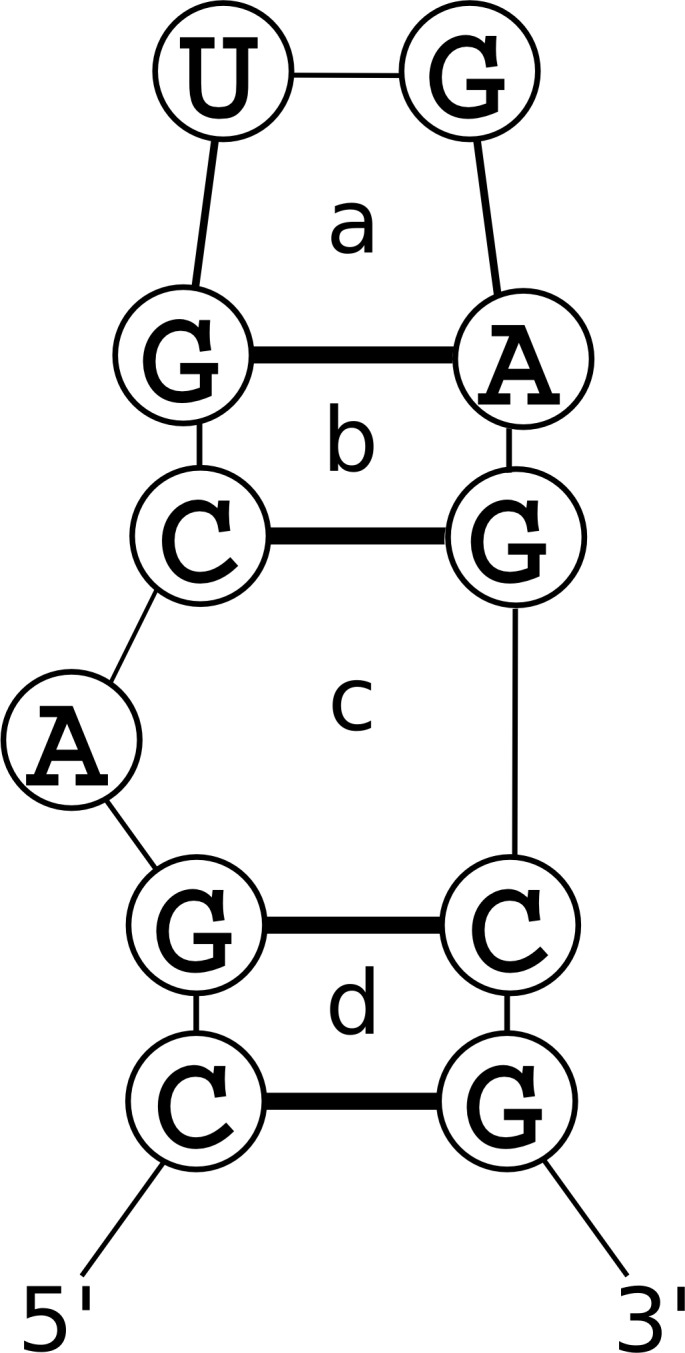
Prediction of RNA tertiary structure from sequence is an important problem, but generating accurate structure models for even short sequences remains difficult. Predictions of RNA tertiary structure tend to be least accurate in loop regions, where non-canonical pairs are important for determining the details of structure. Non-canonical pairs can be predicted using a knowledge-based model of structure that scores nucleotide cyclic motifs, or NCMs. In this work, a partition function algorithm is introduced that allows the estimation of base pairing probabilities for both canonical and non-canonical interactions. Pairs that are predicted to be probable are more likely to be found in the true structure than pairs of lower probability. Pair probability estimates can be further improved by predicting the structure conserved across multiple homologous sequences using the TurboFold algorithm. These pairing probabilities, used in concert with prior knowledge of the canonical secondary structure, allow accurate inference of non-canonical pairs, an important step towards accurate prediction of the full tertiary structure. Software to predict non-canonical base pairs and pairing probabilities is now provided as part of the RNAstructure software package.
从序列预测RNA三级结构是一个重要问题,但即使是短序列生成准确的结构模型仍然困难。RNA三级结构预测在环区往往最不准确,在环区非经典碱基对对于确定结构细节很重要。非经典碱基对可以使用基于结构知识的模型进行预测,该模型对核苷酸环状基序(或NCM)进行评分。在这项工作中,引入了一种配分函数算法,该算法允许估计经典和非经典相互作用的碱基配对概率。预测可能形成的碱基对比概率较低的碱基对更有可能出现在真实结构中。通过使用TurboFold算法预测多个同源序列中保守的结构,可以进一步提高配对概率估计。这些配对概率与经典二级结构的先验知识一起使用,能够准确推断非经典碱基对,这是朝着准确预测完整三级结构迈出的重要一步。现在,作为RNAstructure软件包的一部分,提供了预测非经典碱基对和配对概率的软件。