Brim Hassan, Yooseph Shibu, Lee Edward, Sherif Zaki A, Abbas Muneer, Laiyemo Adeyinka O, Varma Sudhir, Torralba Manolito, Dowd Scot E, Nelson Karen E, Pathmasiri Wimal, Sumner Susan, de Vos Willem, Liang Qiaoyi, Yu Jun, Zoetendal Erwin, Ashktorab Hassan
Department of Pathology and Department of Medicine, Microbiology and Cancer Center, College of Medicine, Howard University, 2041 Georgia Avenue, Washington, DC 20060, USA.
J. Craig Venter Institute, La Jolla, CA 92037, USA.
Genes (Basel). 2017 Nov 9;8(11):314. doi: 10.3390/genes8110314.
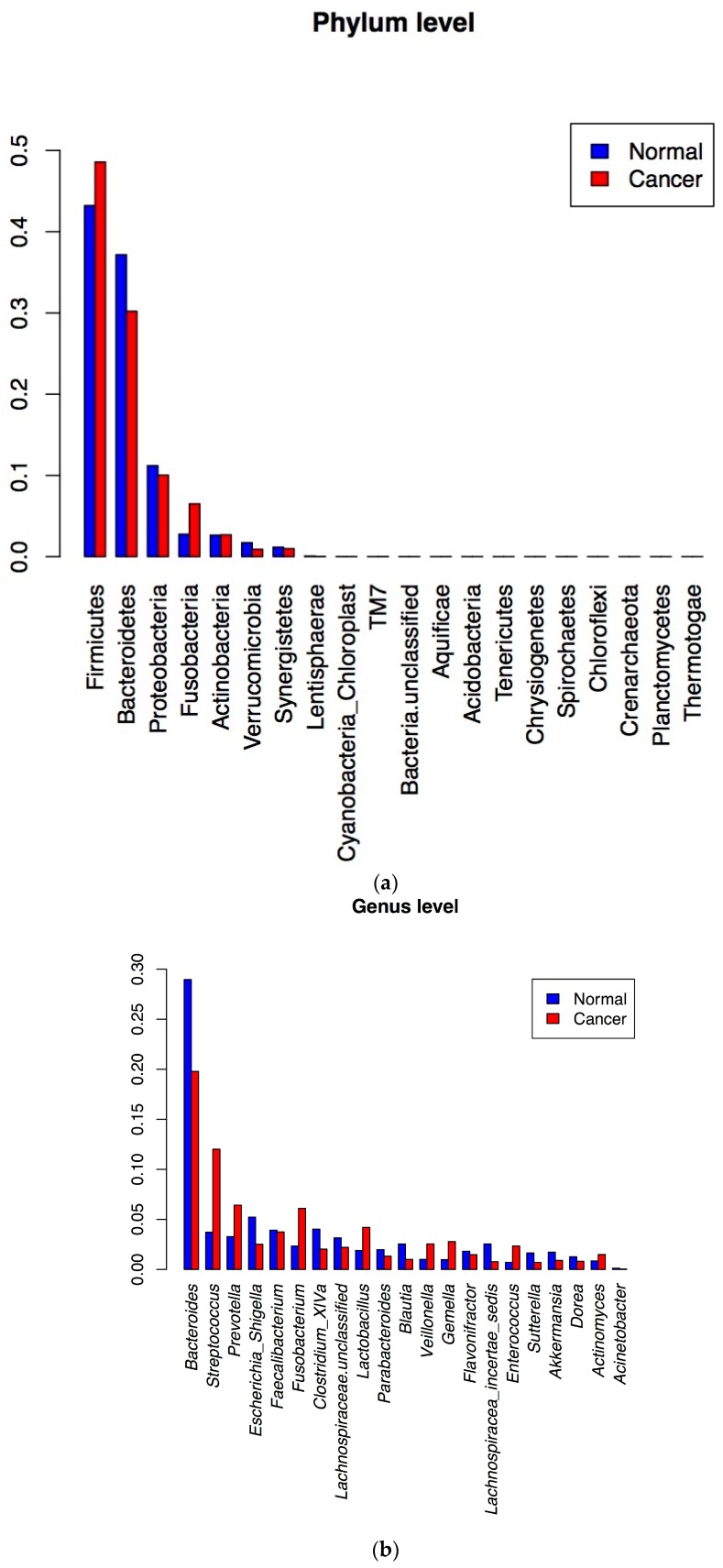
Increasing evidence suggests a role of the gut microbiota in colorectal carcinogenesis (CRC). To detect bacterial markers of colorectal cancer in African Americans a metabolomic analysis was performed on fecal water extracts. DNA from stool samples of adenoma and healthy subjects and from colon cancer and matched normal tissues was analyzed to determine the microbiota composition (using 16S rDNA) and genomic content (metagenomics). Metagenomic functions with discriminative power between healthy and neoplastic specimens were established. Quantitative Polymerase Chain Reaction (q-PCR) using primers and probes specific to sp. VT_162 were used to validate this bacterium association with neoplastic transformation in stool samples from two independent cohorts of African Americans and Chinese patients with colorectal lesions. The metabolomic analysis of adenomas revealed low amino acids content. The microbiota in both cancer vs. normal tissues and adenoma vs. normal stool samples were different at the 16S rRNA gene level. Cross-mapping of metagenomic data led to 9 markers with significant discriminative power between normal and diseased specimens. These markers identified with sp. VT_162. Q-PCR data showed a statistically significant presence of this bacterium in advanced adenoma and cancer samples in an independent cohort of CRC patients. We defined metagenomic functions from sp. VT_162 with discriminative power among cancers vs. matched normal and adenomas vs. healthy subjects' stools. sp. VT_162 specific 16S rDNA was validated in an independent cohort. These findings might facilitate non-invasive screening for colorectal cancer.
越来越多的证据表明肠道微生物群在结直肠癌(CRC)发生中起作用。为了检测非裔美国人结直肠癌的细菌标志物,对粪便水提取物进行了代谢组学分析。分析腺瘤和健康受试者以及结肠癌和匹配的正常组织的粪便样本中的DNA,以确定微生物群组成(使用16S rDNA)和基因组含量(宏基因组学)。建立了在健康和肿瘤标本之间具有判别能力的宏基因组功能。使用针对sp. VT_162的特异性引物和探针进行定量聚合酶链反应(q-PCR),以验证这种细菌与来自两个独立队列的非裔美国人和中国结直肠病变患者粪便样本中肿瘤转化的关联。腺瘤的代谢组学分析显示氨基酸含量低。在16S rRNA基因水平上,癌组织与正常组织以及腺瘤与正常粪便样本中的微生物群均不同。宏基因组数据的交叉映射产生了9个在正常和患病标本之间具有显著判别能力的标志物。这些标志物与sp. VT_162相关。q-PCR数据显示,在一个独立的CRC患者队列中,该细菌在晚期腺瘤和癌症样本中具有统计学意义的存在。我们定义了sp. VT_162的宏基因组功能,其在癌组织与匹配的正常组织以及腺瘤与健康受试者粪便之间具有判别能力。sp. VT_162特异性16S rDNA在一个独立队列中得到验证。这些发现可能有助于结直肠癌的非侵入性筛查。