College of Pharmacy, Western University of Health Sciences , Pomona, California 91766, United States.
Graduate College of Biomedical Sciences, Western University of Health Sciences , Pomona, California 91766, United States.
J Am Chem Soc. 2017 Dec 13;139(49):17945-17952. doi: 10.1021/jacs.7b08938. Epub 2017 Nov 29.
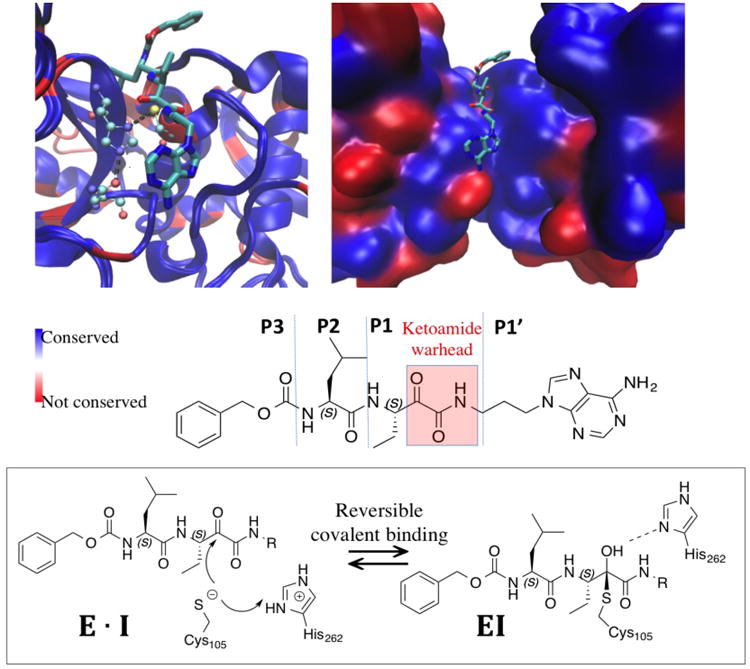
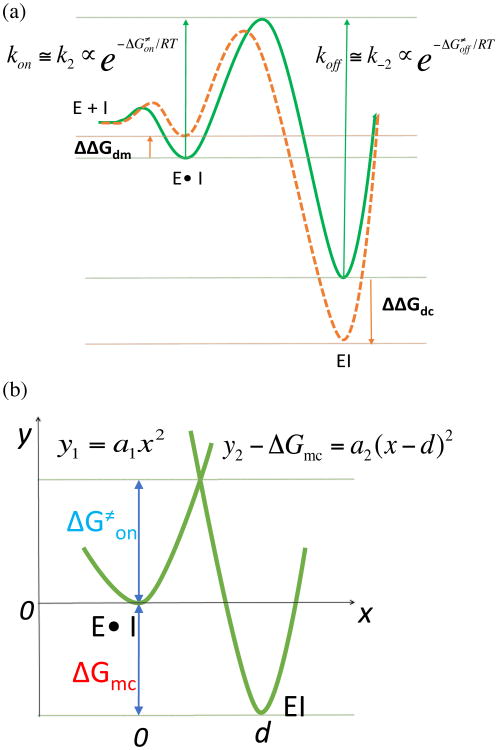
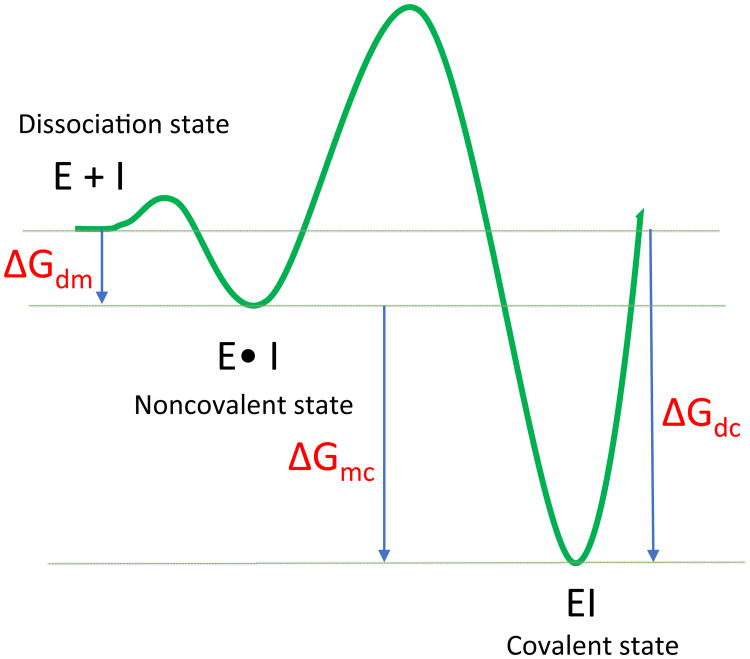
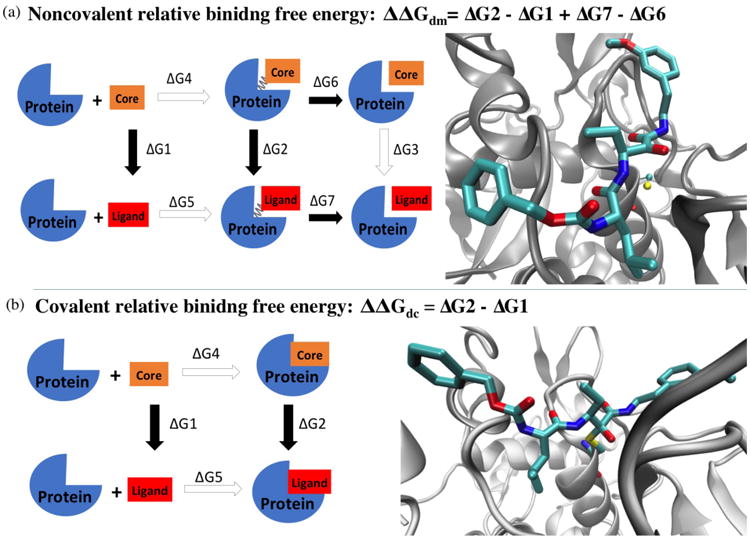
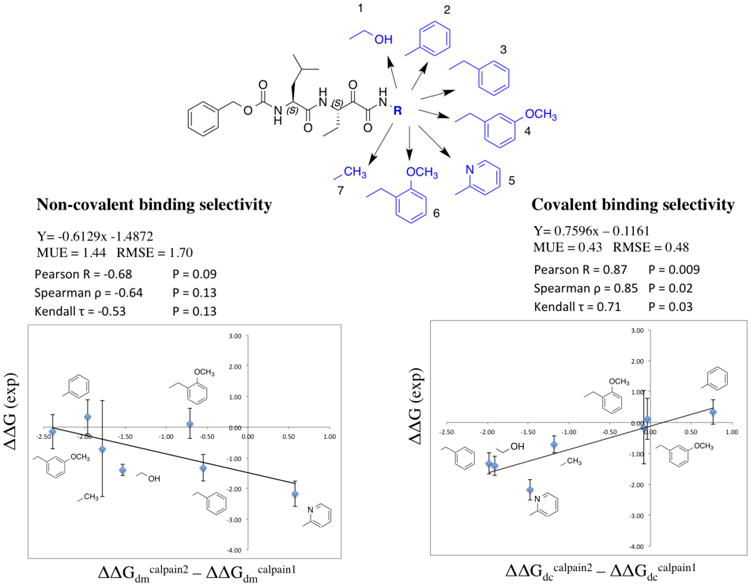
Reversible covalent inhibitors have many clinical advantages over noncovalent or irreversible covalent drugs. However, apart from selecting a warhead, substantial efforts in design and synthesis are needed to optimize noncovalent interactions to improve target-selective binding. Computational prediction of binding affinity for reversible covalent inhibitors presents a unique challenge since the binding process consists of multiple steps, which are not necessarily independent of each other. In this study, we lay out the relation between relative binding free energy and the overall reversible covalent binding affinity using a two-state binding model. To prove the concept, we employed free energy perturbation (FEP) coupled with λ-exchange molecular dynamics method to calculate the binding free energy of a series of α-ketoamide analogues relative to a common warhead scaffold, in both noncovalent and covalent binding states, and for two highly homologous proteases, calpain-1 and calpain-2. We conclude that covalent binding state alone, in general, can be used to predict reversible covalent binding selectivity. However, exceptions may exist. Therefore, we also discuss the conditions under which the noncovalent binding step is no longer negligible and propose to combine the relative FEP calculations with a single QM/MM calculation of warhead to predict the binding affinity and binding kinetics. Our FEP calculations also revealed that covalent and noncovalent binding states of an inhibitor do not necessarily exhibit the same selectivity. Thus, investigating both binding states, as well as the kinetics will provide extremely useful information for optimizing reversible covalent inhibitors.
可逆共价抑制剂相对于非共价或不可逆共价药物具有许多临床优势。然而,除了选择弹头外,还需要进行大量的设计和合成工作,以优化非共价相互作用,提高靶标选择性结合。由于结合过程包含多个步骤,这些步骤不一定相互独立,因此预测可逆共价抑制剂的结合亲和力具有独特的挑战。在这项研究中,我们使用两态结合模型来阐述相对结合自由能与整体可逆共价结合亲和力之间的关系。为了证明这一概念,我们采用自由能微扰(FEP)结合 λ 交换分子动力学方法,计算了一系列 α-酮酰胺类似物相对于常见弹头支架的结合自由能,这些类似物在非共价和共价结合状态下,针对两种高度同源的蛋白酶,钙蛋白酶-1 和钙蛋白酶-2。我们得出结论,一般来说,共价结合状态本身可用于预测可逆共价结合选择性。但是,可能存在例外。因此,我们还讨论了非共价结合步骤不再可以忽略的条件,并提出将相对 FEP 计算与弹头的单个 QM/MM 计算相结合,以预测结合亲和力和结合动力学。我们的 FEP 计算还表明,抑制剂的共价和非共价结合状态不一定表现出相同的选择性。因此,研究两种结合状态以及动力学将为优化可逆共价抑制剂提供非常有用的信息。