Nichio Bruno T L, Marchaukoski Jeroniza Nunes, Raittz Roberto Tadeu
Department of Bioinformatics, Professional and Technical Education Sector, Federal University of Paraná, Curitiba, Brazil.
Front Genet. 2017 Oct 31;8:165. doi: 10.3389/fgene.2017.00165. eCollection 2017.
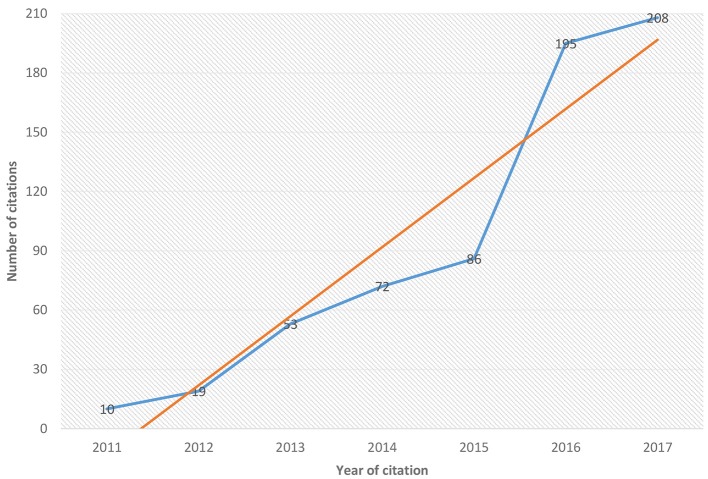
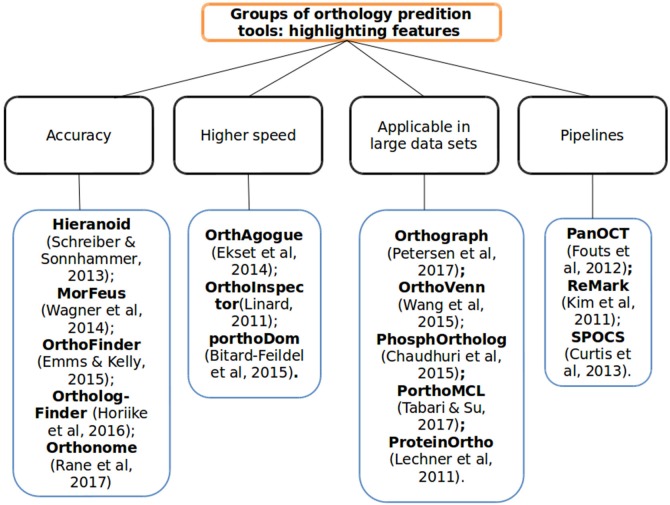
Nowadays defying homology relationships among sequences is essential for biological research. Within homology the analysis of orthologs sequences is of great importance for computational biology, annotation of genomes and for phylogenetic inference. Since 2007, with the increase in the number of new sequences being deposited in large biological databases, researchers have begun to analyse computerized methodologies and tools aimed at selecting the most promising ones in the prediction of orthologous groups. Literature in this field of research describes the problems that the majority of available tools show, such as those encountered in accuracy, time required for analysis (especially in light of the increasing volume of data being submitted, which require faster techniques) and the automatization of the process without requiring manual intervention. Conducting our search through BMC, Google Scholar, NCBI PubMed, and Expasy, we examined more than 600 articles pursuing the most recent techniques and tools developed to solve most the problems still existing in orthology detection. We listed the main computational tools created and developed between 2011 and 2017, taking into consideration the differences in the type of orthology analysis, outlining the main features of each tool and pointing to the problems that each one tries to address. We also observed that several tools still use as their main algorithm the BLAST "all-against-all" methodology, which entails some limitations, such as limited number of queries, computational cost, and high processing time to complete the analysis. However, new promising tools are being developed, like OrthoVenn (which uses the Venn diagram to show the relationship of ortholog groups generated by its algorithm); or proteinOrtho (which improves the accuracy of ortholog groups); or ReMark (tackling the integration of the pipeline to turn the entry process automatic); or OrthAgogue (using algorithms developed to minimize processing time); and proteinOrtho (developed for dealing with large amounts of biological data). We made a comparison among the main features of four tool and tested them using four for prokaryotic genomas. We hope that our review can be useful for researchers and will help them in selecting the most appropriate tool for their work in the field of orthology.
如今,突破序列间的同源关系对生物学研究至关重要。在同源性范畴内,直系同源序列分析对计算生物学、基因组注释及系统发育推断具有重大意义。自2007年以来,随着大量新序列存入大型生物数据库,研究人员开始分析旨在挑选出预测直系同源组中最具潜力工具的计算机化方法和工具。该研究领域的文献描述了大多数现有工具存在的问题,比如准确性方面遇到的问题、分析所需时间(尤其是鉴于提交的数据量不断增加,这需要更快的技术)以及无需人工干预的流程自动化问题。我们通过生物医学中心(BMC)、谷歌学术、美国国立医学图书馆国立生物技术信息中心(NCBI)的PubMed和专家蛋白质分析系统(Expasy)进行搜索,查阅了600多篇文章,探寻为解决直系同源性检测中仍然存在的大多数问题而开发的最新技术和工具。我们列出了2011年至2017年间创建和开发的主要计算工具,考虑到直系同源性分析类型的差异,概述了每个工具的主要特征,并指出每个工具试图解决的问题。我们还观察到,一些工具仍将BLAST“全对全”方法作为其主要算法,这存在一些局限性,如查询数量有限、计算成本高以及完成分析所需的处理时间长。然而,正在开发新的有前景的工具,如OrthoVenn(它使用维恩图展示其算法生成的直系同源组关系);或ProteinOrtho(它提高了直系同源组的准确性);或ReMark(解决流程整合问题以使输入过程自动化);或OrthoAgogue(使用为最小化处理时间而开发的算法);以及ProteinOrtho(为处理大量生物数据而开发)。我们对四种工具的主要特征进行了比较,并使用四个原核基因组对它们进行了测试。我们希望我们的综述对研究人员有用,并将帮助他们在直系同源性领域的工作中选择最合适的工具。