Animal Breeding and Genomics Centre, Wageningen UR Livestock Research, Lelystad, The Netherlands.
Genet Sel Evol. 2011 Jul 5;43(1):26. doi: 10.1186/1297-9686-43-26.
Genomic selection has become a very important tool in animal genetics and is rapidly emerging in plant genetics. It holds the promise to be particularly beneficial to select for traits that are difficult or expensive to measure, such as traits that are measured in one environment and selected for in another environment. The objective of this paper was to develop three models that would permit multi-trait genomic selection by combining scarcely recorded traits with genetically correlated indicator traits, and to compare their performance to single-trait models, using simulated datasets.
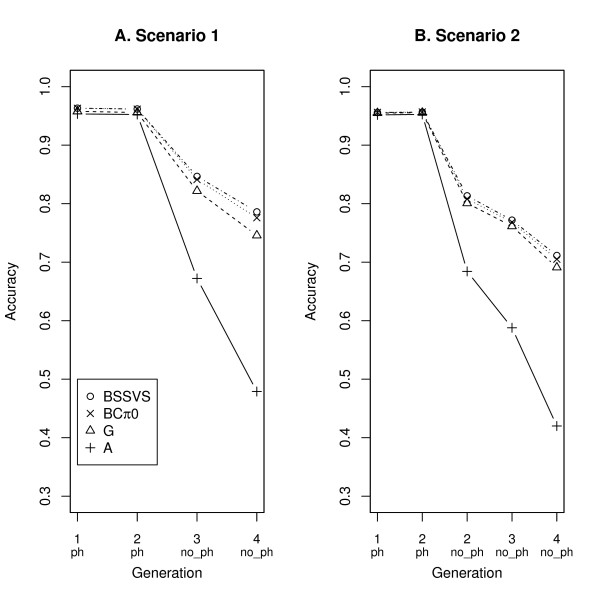
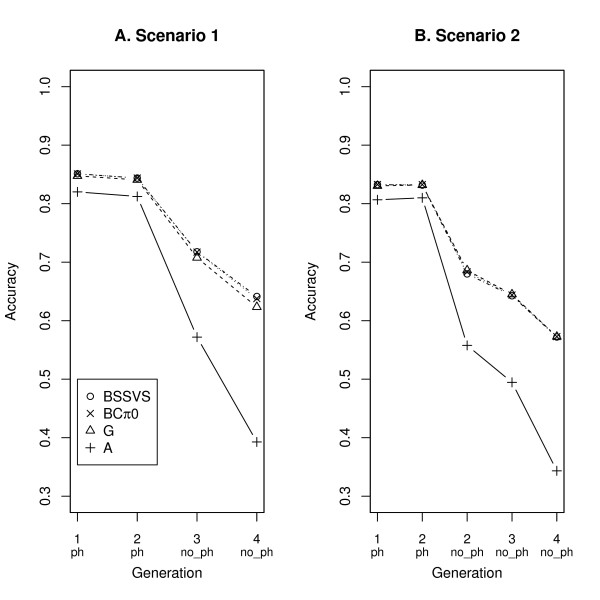
Three (SNP) Single Nucleotide Polymorphism based models were used. Model G and BCπ0 assumed that contributed (co)variances of all SNP are equal. Model BSSVS sampled SNP effects from a distribution with large (or small) effects to model SNP that are (or not) associated with a quantitative trait locus. For reasons of comparison, model A including pedigree but not SNP information was fitted as well.
In terms of accuracies for animals without phenotypes, the models generally ranked as follows: BSSVS > BCπ0 > G > > A. Using multi-trait SNP-based models, the accuracy for juvenile animals without any phenotypes increased up to 0.10. For animals with phenotypes on an indicator trait only, accuracy increased up to 0.03 and 0.14, for genetic correlations with the evaluated trait of 0.25 and 0.75, respectively.
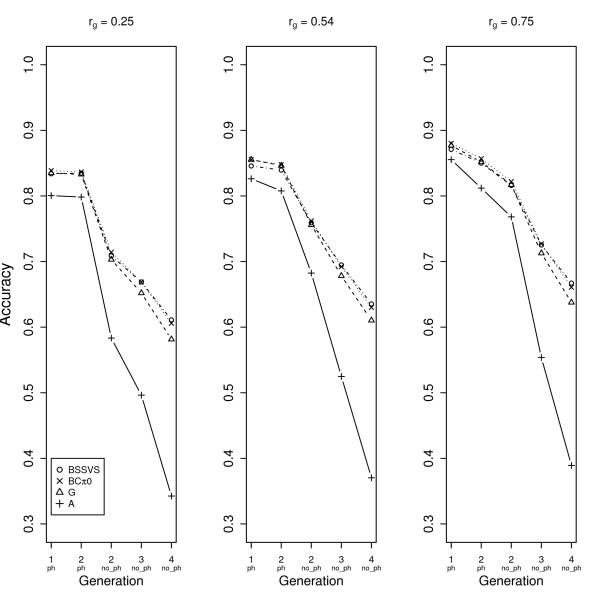
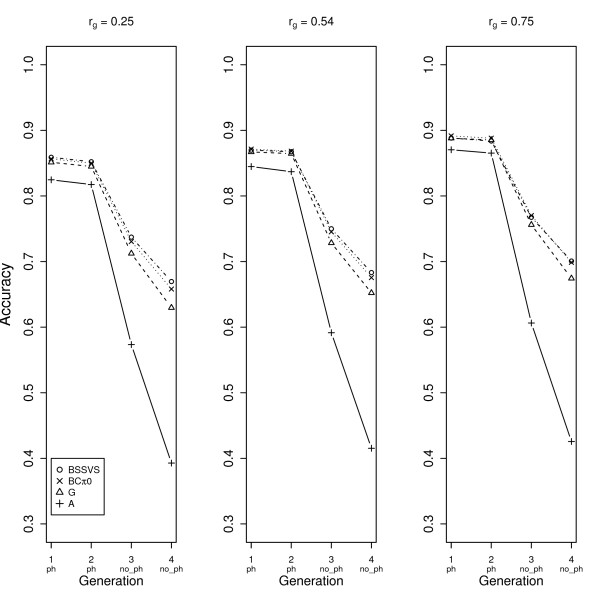
When the indicator trait had a genetic correlation lower than 0.5 with the trait of interest in our simulated data, the accuracy was higher if genotypes rather than phenotypes were obtained for the indicator trait. However, when genetic correlations were higher than 0.5, using an indicator trait led to higher accuracies for selection candidates. For different combinations of traits, the level of genetic correlation below which genotyping selection candidates is more effective than obtaining phenotypes for an indicator trait, needs to be derived considering at least the heritabilities and the numbers of animals recorded for the traits involved.
基因组选择已成为动物遗传学中非常重要的工具,并且在植物遗传学中也迅速兴起。它有望特别有益于选择那些难以或昂贵测量的性状,例如在一种环境中测量并在另一种环境中选择的性状。本文的目的是开发三种模型,通过将记录甚少的性状与遗传相关的指示性状相结合,实现多性状基因组选择,并使用模拟数据集比较它们的性能与单性状模型。
使用了三种基于单核苷酸多态性(SNP)的模型。模型 G 和 BCπ0 假设所有 SNP 的贡献(协)方差相等。模型 BSSVS 从具有大(或小)效应的分布中抽样 SNP 效应,以模拟与数量性状基因座相关(或不相关)的 SNP。出于比较的原因,还拟合了包含系谱但不包含 SNP 信息的模型 A。
就无表型动物的准确性而言,这些模型通常排名如下:BSSVS > BCπ0 > G > > A。使用多性状 SNP 基模型,无任何表型的幼年动物的准确性最高可达 0.10。对于仅在指示性状上具有表型的动物,对于与评估性状的遗传相关系数分别为 0.25 和 0.75,准确性最高分别可达 0.03 和 0.14。
在我们模拟数据中,当指示性状与感兴趣性状的遗传相关性低于 0.5 时,如果获得指示性状的基因型而不是表型,则准确性更高。然而,当遗传相关性高于 0.5 时,使用指示性状可以提高选择候选者的准确性。对于不同性状的组合,需要考虑至少性状的遗传力和记录的动物数量,得出基因型选择候选者比获得指示性状的表型更有效的遗传相关水平。