Scelfo Chiara, Caminati Antonella, Harari Sergio
Unità Operativa di Pneumologia e Terapia Semi-Intensiva Respiratoria, Servizio di Fisiopatologia Respiratoria ed Emodinamica Polmonare, Ospedale San Giuseppe, Multimedica IRCCS, Milan, Italy.
F1000Res. 2017 Nov 27;6:2052. doi: 10.12688/f1000research.10720.1. eCollection 2017.
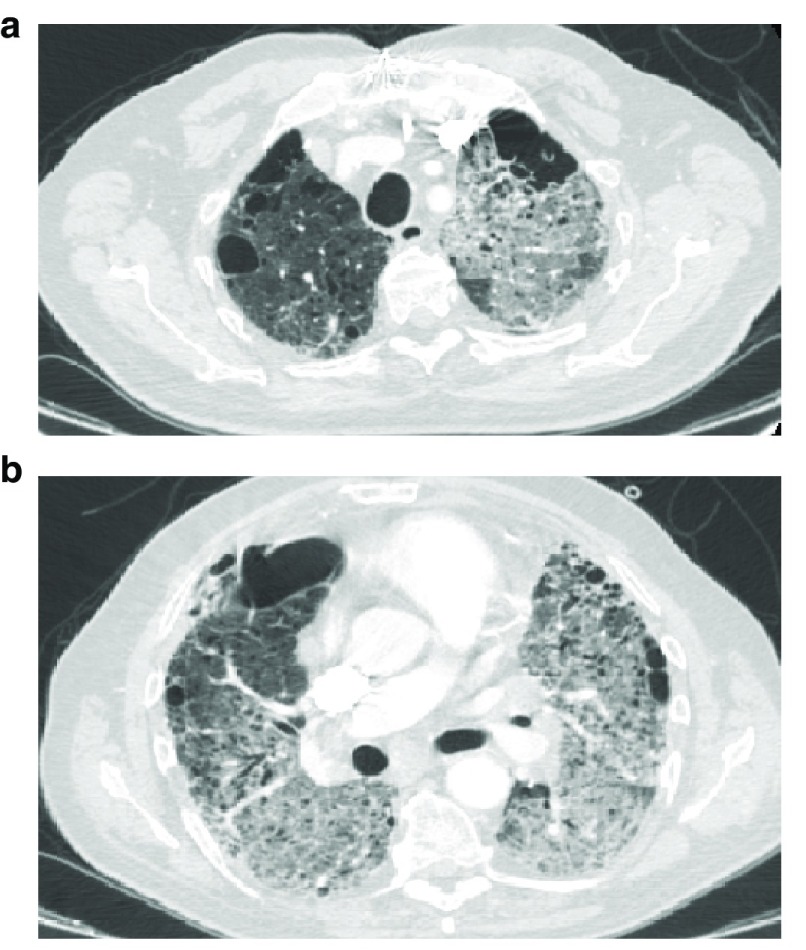
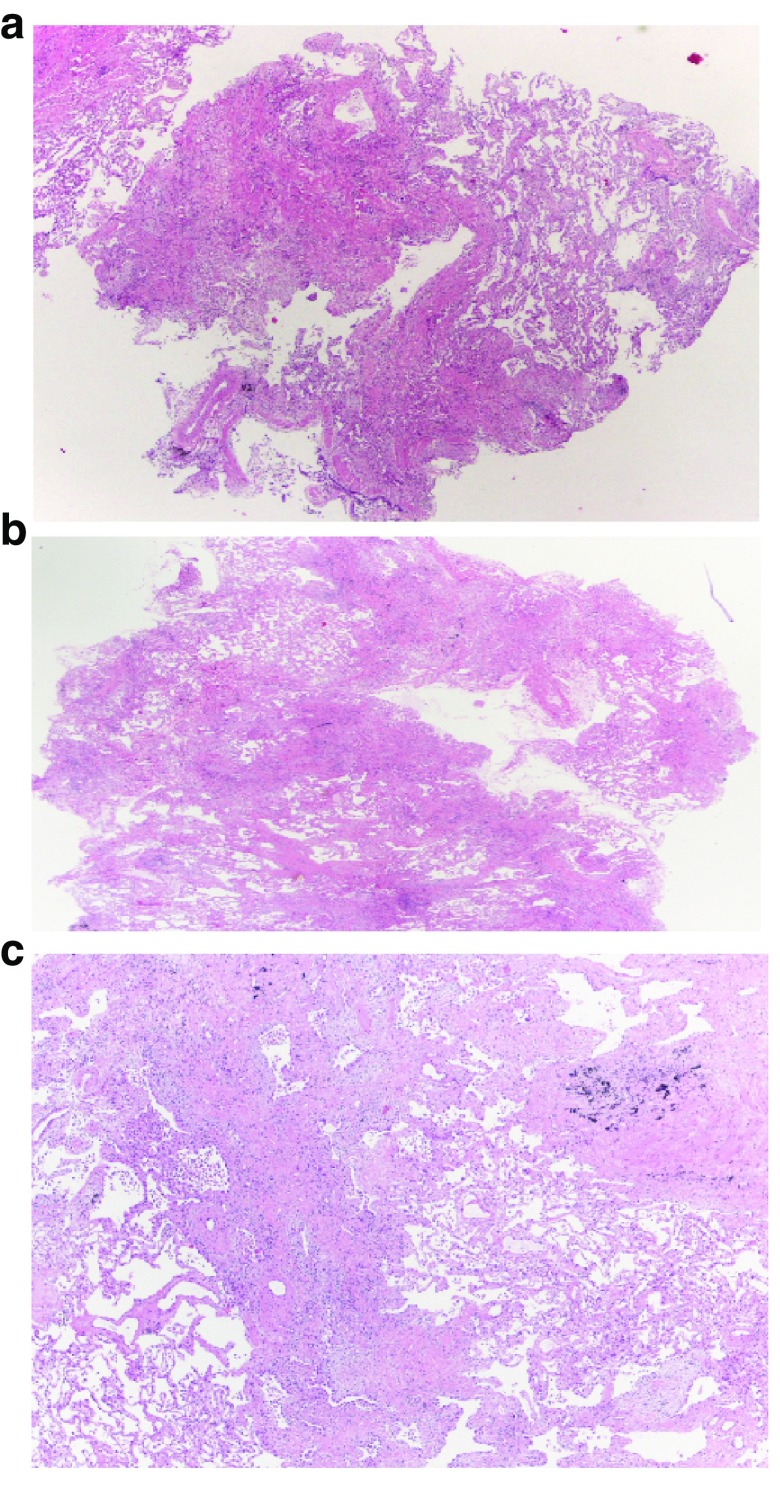
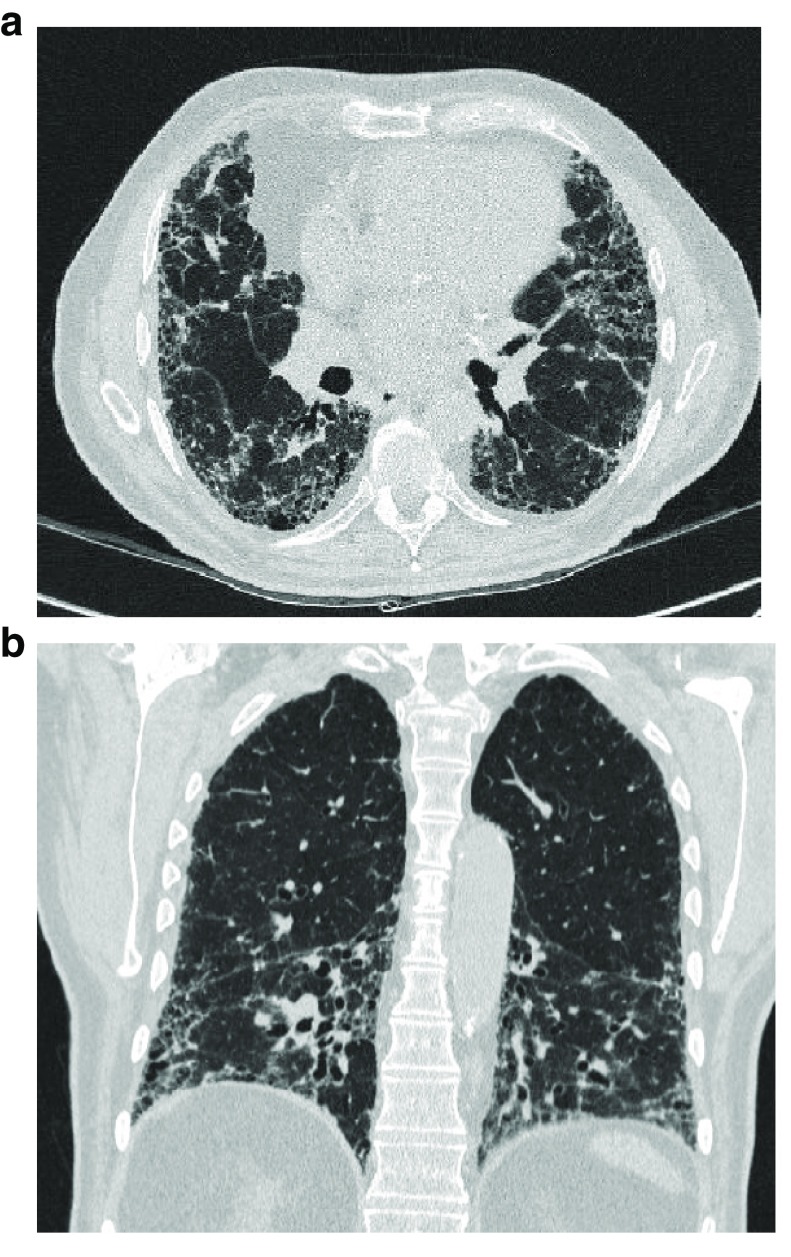
Idiopathic pulmonary fibrosis (IPF) is a rare pulmonary disease with a poor prognosis and severe impact on quality of life. Early diagnosis is still challenging and important delays are registered before final diagnosis can be reached. Available tools fail to predict the variable course of the disease and to evaluate response to antifibrotic drugs. Despite the recent approval of pirfenidone and nintedanib, significant challenges remain to improve prognosis and quality of life. It is hoped that the new insights gained in pathobiology in the last few years will lead to further advances in the diagnosis and management of IPF. Currently, early diagnosis and prompt initiation of treatments reducing lung function loss offer the best hope for improved outcomes. This article aims at providing an overview of recent advances in managing patients with IPF and has a particular focus on how to reach a diagnosis, manage comorbidities and lung transplantation, care for the non-pharmacological needs of patients, and address palliative care.
特发性肺纤维化(IPF)是一种罕见的肺部疾病,预后较差,对生活质量有严重影响。早期诊断仍然具有挑战性,在最终确诊之前会出现显著延误。现有的工具无法预测疾病的多变进程,也无法评估对抗纤维化药物的反应。尽管吡非尼酮和尼达尼布最近已获批准,但在改善预后和生活质量方面仍存在重大挑战。希望过去几年在病理生物学方面获得的新见解将推动IPF诊断和管理的进一步进展。目前,早期诊断并迅速启动减少肺功能丧失的治疗为改善预后带来了最大希望。本文旨在概述IPF患者管理的最新进展,特别关注如何进行诊断、管理合并症和肺移植、满足患者的非药物需求以及提供姑息治疗。