Institute of Computing Science, Poznan University of Technology, Poznan 60-965, Poland.
Institute of Bioorganic Chemistry, Polish Academy of Sciences, Poznan 61-704, Poland.
Bioinformatics. 2018 Apr 15;34(8):1304-1312. doi: 10.1093/bioinformatics/btx783.
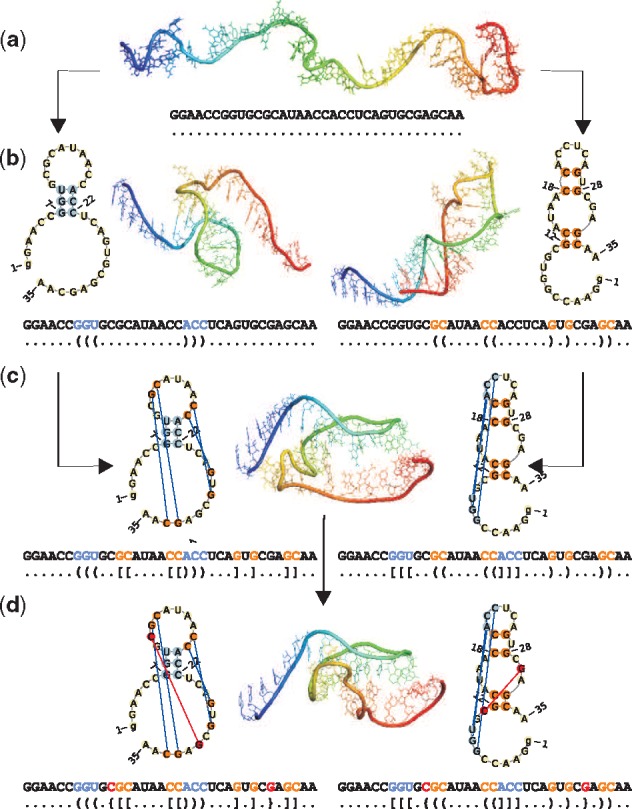
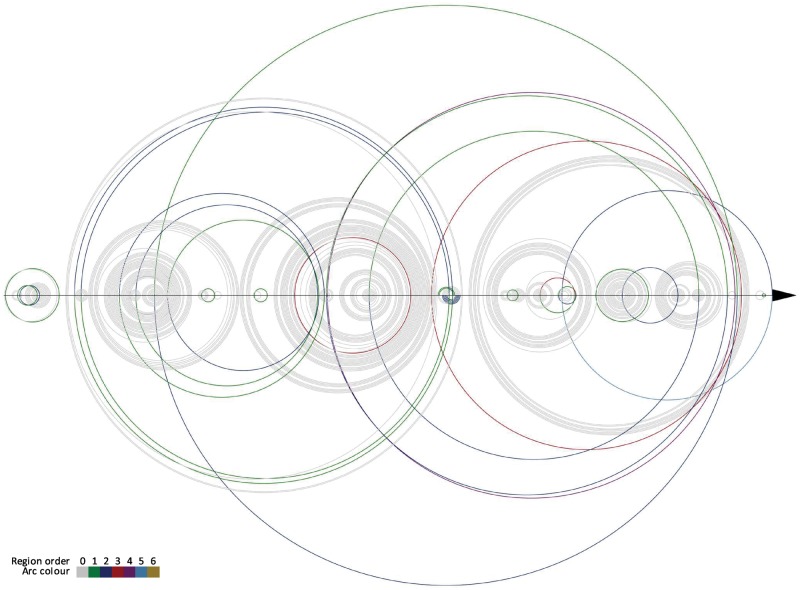
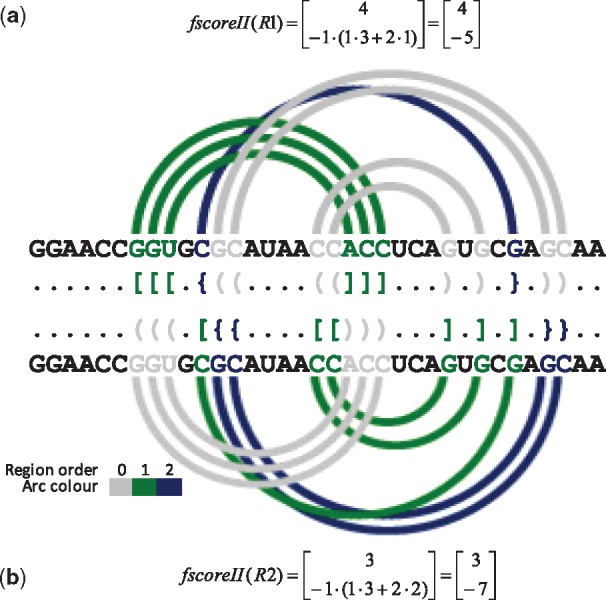
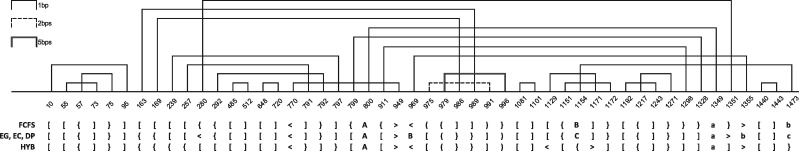
Understanding the formation, architecture and roles of pseudoknots in RNA structures are one of the most difficult challenges in RNA computational biology and structural bioinformatics. Methods predicting pseudoknots typically perform this with poor accuracy, often despite experimental data incorporation. Existing bioinformatic approaches differ in terms of pseudoknots' recognition and revealing their nature. A few ways of pseudoknot classification exist, most common ones refer to a genus or order. Following the latter one, we propose new algorithms that identify pseudoknots in RNA structure provided in BPSEQ format, determine their order and encode in dot-bracket-letter notation. The proposed encoding aims to illustrate the hierarchy of RNA folding.
New algorithms are based on dynamic programming and hybrid (combining exhaustive search and random walk) approaches. They evolved from elementary algorithm implemented within the workflow of RNA FRABASE 1.0, our database of RNA structure fragments. They use different scoring functions to rank dissimilar dot-bracket representations of RNA structure. Computational experiments show an advantage of new methods over the others, especially for large RNA structures.
Presented algorithms have been implemented as new functionality of RNApdbee webserver and are ready to use at http://rnapdbee.cs.put.poznan.pl.
Supplementary data are available at Bioinformatics online.
理解 RNA 结构中假结的形成、结构和作用是 RNA 计算生物学和结构生物信息学中最具挑战性的问题之一。预测假结的方法通常准确性较差,尽管纳入了实验数据。现有的生物信息学方法在识别假结和揭示其性质方面存在差异。存在几种假结分类方法,最常见的方法是指属或阶。按照后者,我们提出了新的算法,用于识别以 BPSEQ 格式提供的 RNA 结构中的假结,确定它们的阶并以点棒字母表示法进行编码。所提出的编码旨在说明 RNA 折叠的层次结构。
新算法基于动态规划和混合(结合穷举搜索和随机游走)方法。它们是从 RNA FRABASE 1.0 工作流程中实现的基本算法演变而来的,RNA FRABASE 1.0 是我们的 RNA 结构片段数据库。它们使用不同的评分函数对 RNA 结构的不同点棒表示进行排序。计算实验表明,新方法优于其他方法,特别是对于大型 RNA 结构。
提出的算法已作为 RNApdbee 网络服务器的新功能实现,并可在 http://rnapdbee.cs.put.poznan.pl 使用。
补充数据可在 Bioinformatics 在线获得。