Mardt Andreas, Pasquali Luca, Wu Hao, Noé Frank
Department of Mathematics and Computer Science, Freie Universität Berlin, Arnimallee 6, 14195, Berlin, Germany.
Nat Commun. 2018 Jan 2;9(1):5. doi: 10.1038/s41467-017-02388-1.
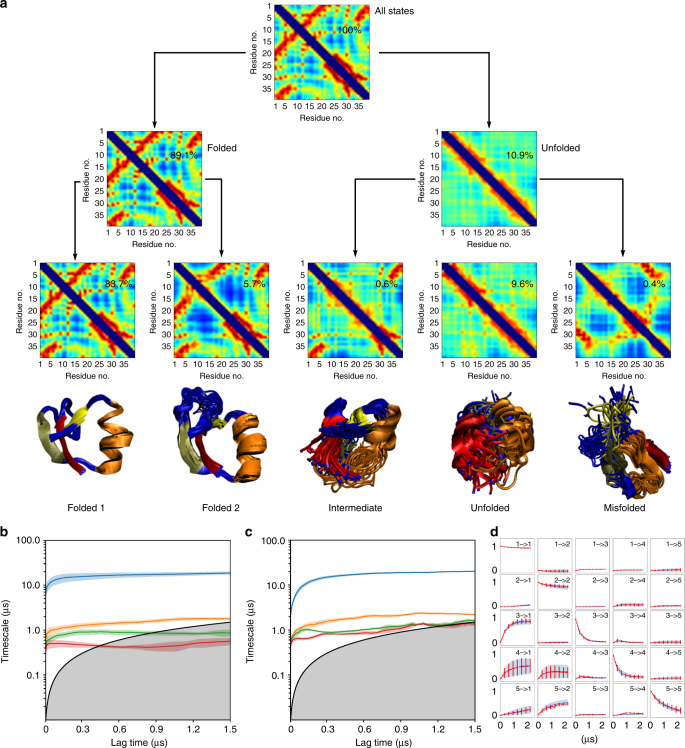
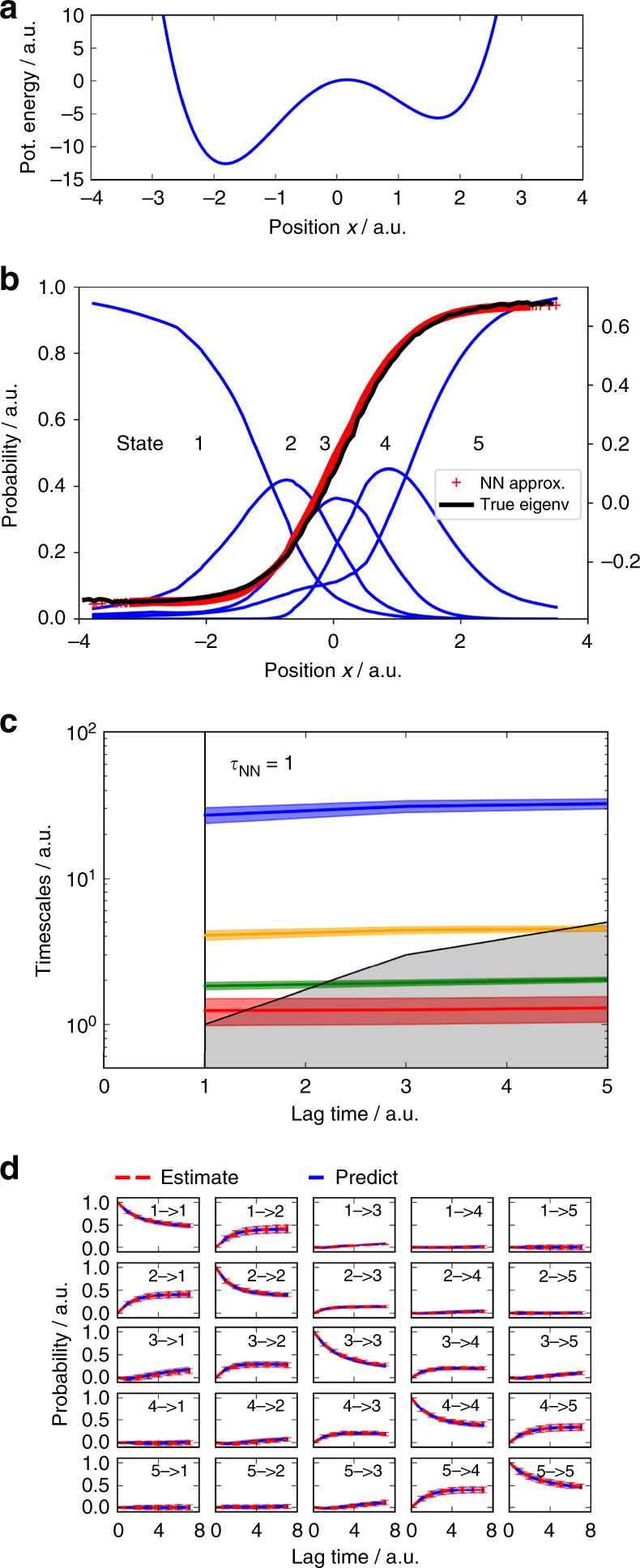
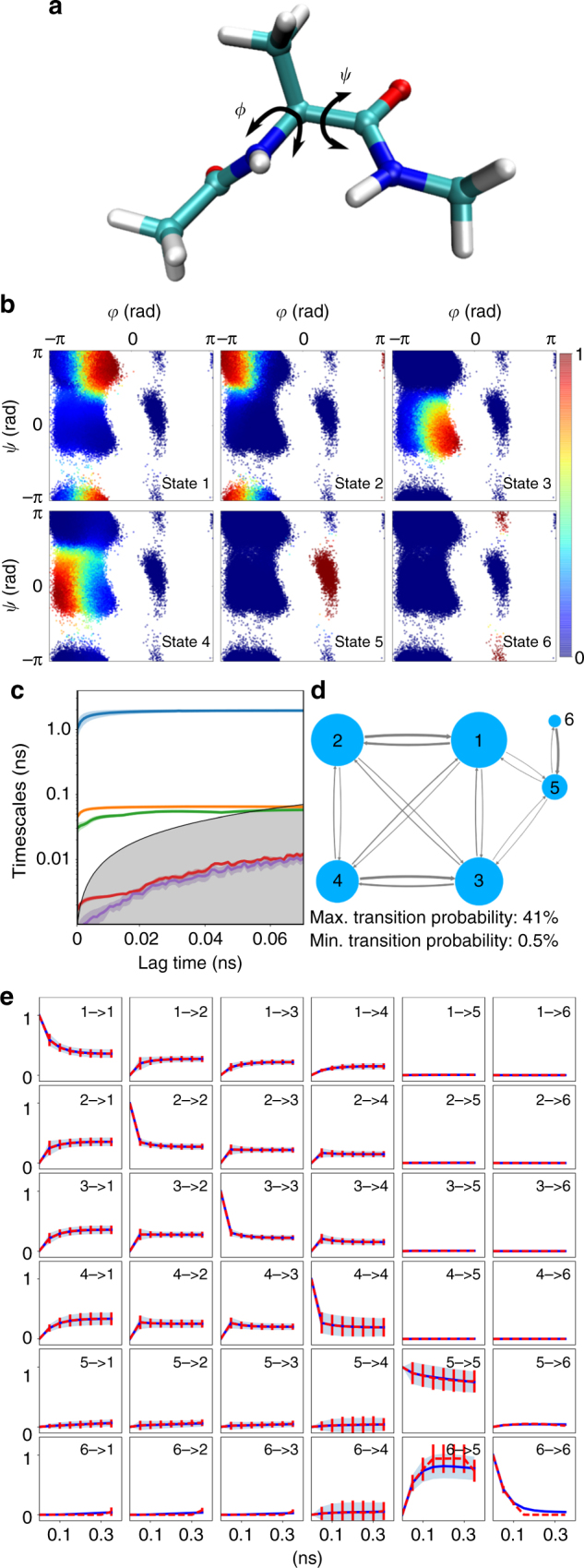
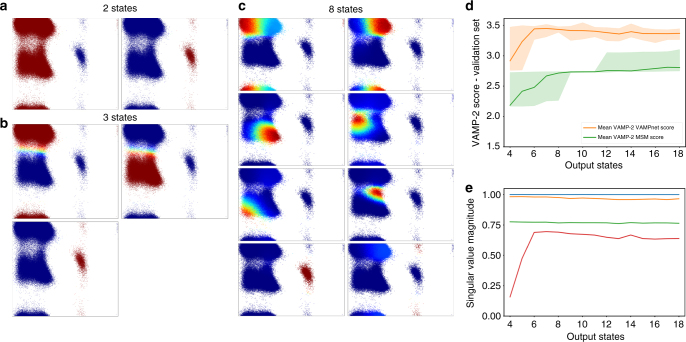
There is an increasing demand for computing the relevant structures, equilibria, and long-timescale kinetics of biomolecular processes, such as protein-drug binding, from high-throughput molecular dynamics simulations. Current methods employ transformation of simulated coordinates into structural features, dimension reduction, clustering the dimension-reduced data, and estimation of a Markov state model or related model of the interconversion rates between molecular structures. This handcrafted approach demands a substantial amount of modeling expertise, as poor decisions at any step will lead to large modeling errors. Here we employ the variational approach for Markov processes (VAMP) to develop a deep learning framework for molecular kinetics using neural networks, dubbed VAMPnets. A VAMPnet encodes the entire mapping from molecular coordinates to Markov states, thus combining the whole data processing pipeline in a single end-to-end framework. Our method performs equally or better than state-of-the-art Markov modeling methods and provides easily interpretable few-state kinetic models.
通过高通量分子动力学模拟来计算生物分子过程(如蛋白质 - 药物结合)的相关结构、平衡和长时间尺度动力学的需求日益增加。当前的方法包括将模拟坐标转换为结构特征、降维、对降维后的数据进行聚类,以及估计马尔可夫状态模型或分子结构间相互转换速率的相关模型。这种手工制作的方法需要大量的建模专业知识,因为任何一步的错误决策都会导致较大的建模误差。在此,我们采用马尔可夫过程变分方法(VAMP)来开发一个使用神经网络的分子动力学深度学习框架,称为VAMPnets。一个VAMPnet对从分子坐标到马尔可夫状态的整个映射进行编码,从而将整个数据处理管道整合在一个端到端的框架中。我们的方法与最先进的马尔可夫建模方法表现相当或更优,并提供易于解释的少状态动力学模型。