Department of Animal Science, North Carolina State University, Raleigh, 27606, NC, USA.
Matatu Inc., 4320 Forest Park Ave., Suite 321, Saint Louis, 63108, MO, USA.
Microbiome. 2018 Jan 4;6(1):4. doi: 10.1186/s40168-017-0384-1.
In pigs, gut bacteria have been shown to play important roles in nutritional, physiological, and immunological processes in the host. However, the contribution of their metagenomes or part of them, which are normally reflected by fragments of 16S rRNA-encoding genes, has yet to be fully investigated.
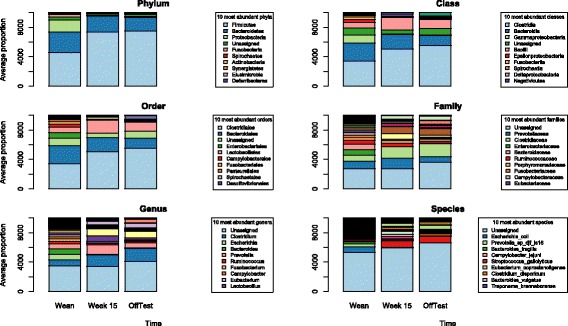
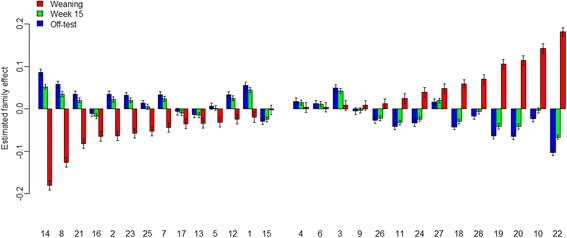
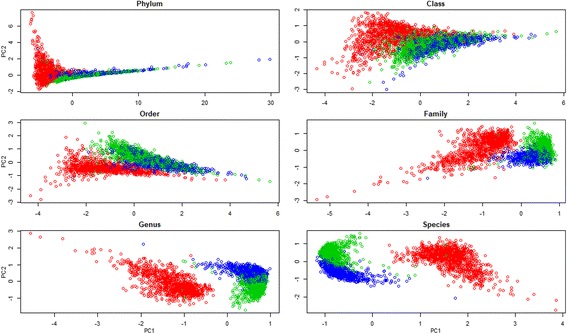
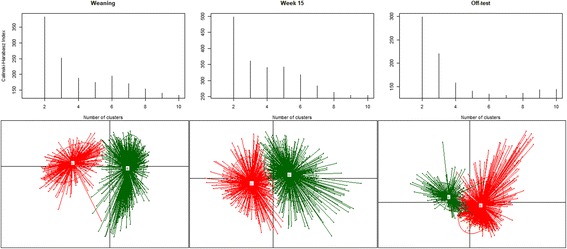
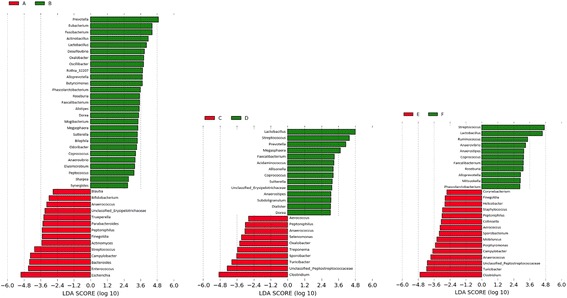
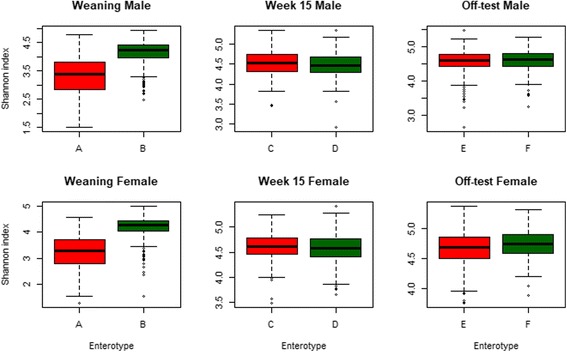
Fecal samples, collected from a population of crossbred pigs at three time points, including weaning, week 15 post weaning (hereafter "week 15"), and end-of-feeding test (hereafter "off-test"), were used to evaluate changes in the composition of the fecal microbiome of each animal over time. This study used 1205, 1295, and 1283 samples collected at weaning, week 15, and off-test, respectively. There were 1039 animals that had samples collected at all three time points and also had phenotypic records on back fat thickness (BF) and average daily body weight gain (ADG). Firmicutes and Bacteroidetes were the most abundant phyla at all three time points. The most abundant genera at all three time points included Clostridium, Escherichia, Bacteroides, Prevotella, Ruminococcus, Fusobacterium, Campylobacter, Eubacterium, and Lactobacillus. Two enterotypes were identified at each time point. However, only enterotypes at week 15 and off-test were significantly associated with BF. We report herein two novel findings: (i) alpha diversity and operational taxonomic unit (OTU) richness were moderately heritable at week 15, h of 0.15 ± 0.06 to 0.16 ± 0.07 and 0.23 ± 0.09 to 0.26 ± 0.08, respectively, as well as at off-test, h of 0.20 ± 0.09 to 0.33 ± 0.10 and 0.17 ± 0.08 to 0.24 ± 0.08, respectively, whereas very low heritability estimates for both measures were detected at weaning; and (ii) alpha diversity at week 15 had strong and negative genetic correlations with BF, - 0.53 ± 0.23 to - 0.45 ± 0.25, as well as with ADG, - 0.53 ± 0.32 to - 0.53 ± 0.29.
These results are important for efforts to genetically improve the domesticated pig because they suggest fecal microbiota diversity can be used as an indicator trait to improve traits that are expensive to measure.
在猪体内,肠道细菌在宿主的营养、生理和免疫过程中发挥着重要作用。然而,它们的宏基因组或其中一部分(通常反映为 16S rRNA 编码基因的片段)的贡献尚未得到充分研究。
本研究从杂交猪群体中采集了三个时间点(断奶、断奶后第 15 周(以下简称“第 15 周”)和饲养结束时(以下简称“结束时”))的粪便样本,以评估每个动物的粪便微生物组组成随时间的变化。本研究分别在断奶、第 15 周和结束时采集了 1205、1295 和 1283 个样本。共有 1039 只动物在三个时间点均采集了样本,并且还记录了背膘厚(BF)和平均日增重(ADG)的表型。厚壁菌门和拟杆菌门在所有三个时间点均为最丰富的门。在所有三个时间点最丰富的菌属包括梭菌属、埃希氏菌属、拟杆菌属、普雷沃氏菌属、真杆菌属、梭杆菌属、弯曲杆菌属、真杆菌属和乳杆菌属。在每个时间点都确定了两种肠型。然而,只有第 15 周和结束时的肠型与 BF 显著相关。我们在此报告两项新发现:(i)第 15 周和结束时的 alpha 多样性和操作分类单元(OTU)丰富度具有中等的遗传力,分别为 0.15±0.06 至 0.16±0.07 和 0.23±0.09 至 0.26±0.08,而断奶时的遗传力估计值非常低;(ii)第 15 周的 alpha 多样性与 BF 呈强负遗传相关,-0.53±0.23 至-0.45±0.25,与 ADG 呈强负遗传相关,-0.53±0.32 至-0.53±0.29。
这些结果对于努力提高家猪的遗传性能非常重要,因为它们表明粪便微生物多样性可用作指示性状,以改善难以测量的性状。