Krewald Vera, Retegan Marius, Cox Nicholas, Messinger Johannes, Lubitz Wolfgang, DeBeer Serena, Neese Frank, Pantazis Dimitrios A
Max Planck Institute for Chemical Energy Conversion , Stiftstr. 34-38 , 45470 Mülheim an der Ruhr , Germany . Email:
Department of Chemistry , Chemical Biological Center (KBC) , Umeå University , 90187 Umeå , Sweden.
Chem Sci. 2015 Mar 1;6(3):1676-1695. doi: 10.1039/c4sc03720k. Epub 2015 Jan 9.
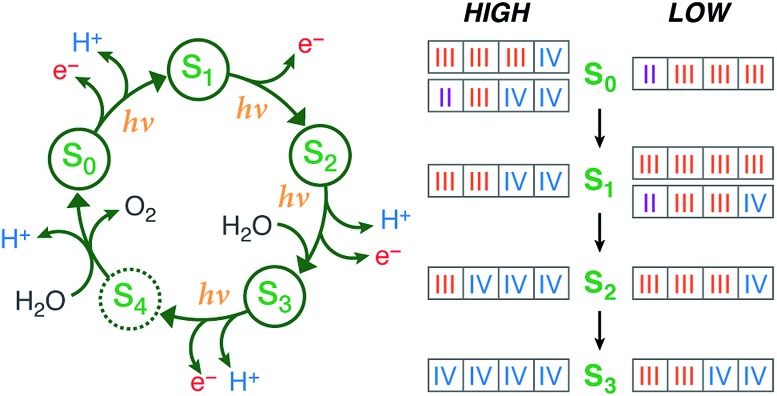
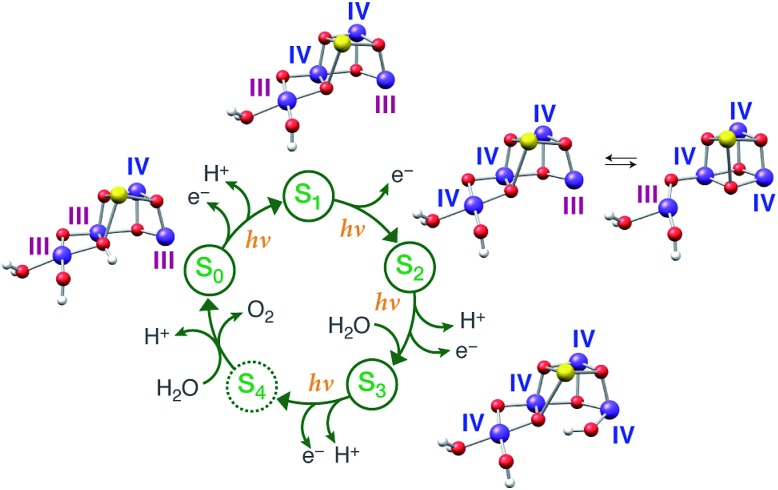
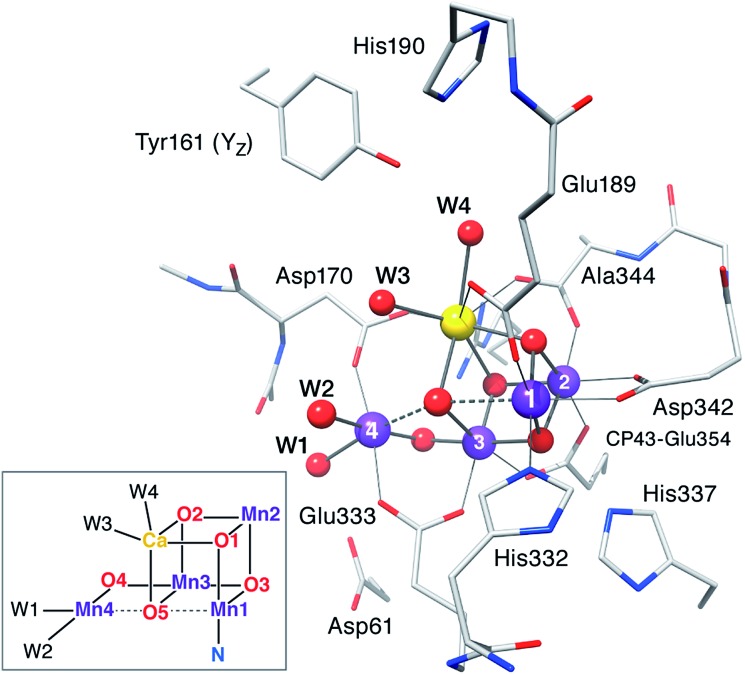
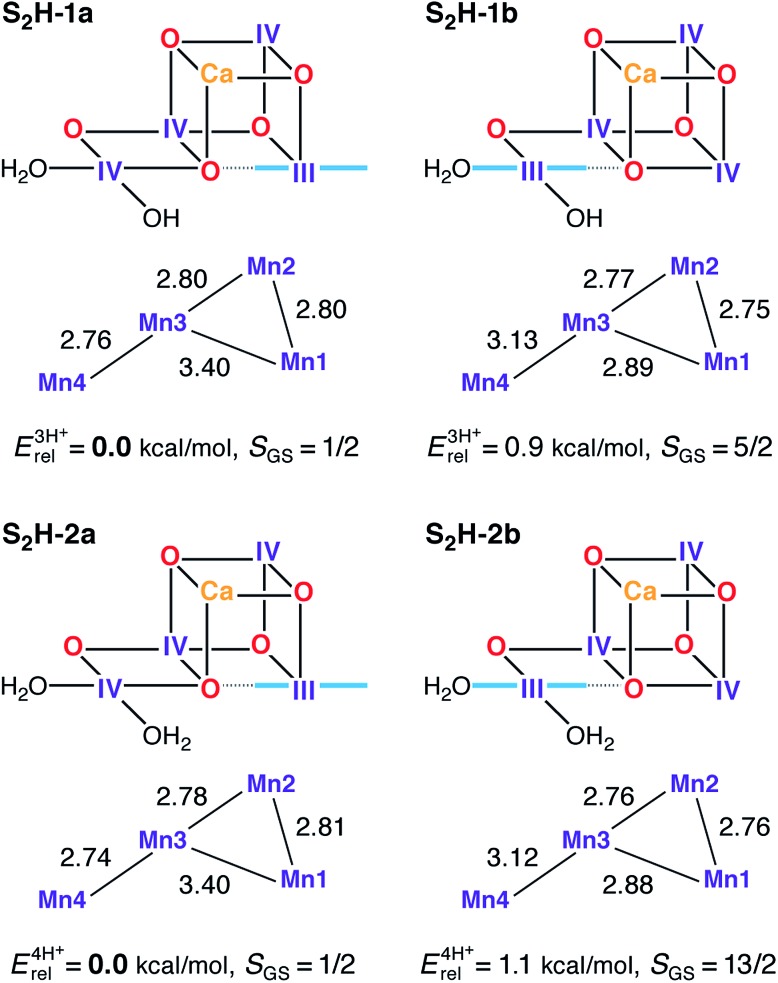
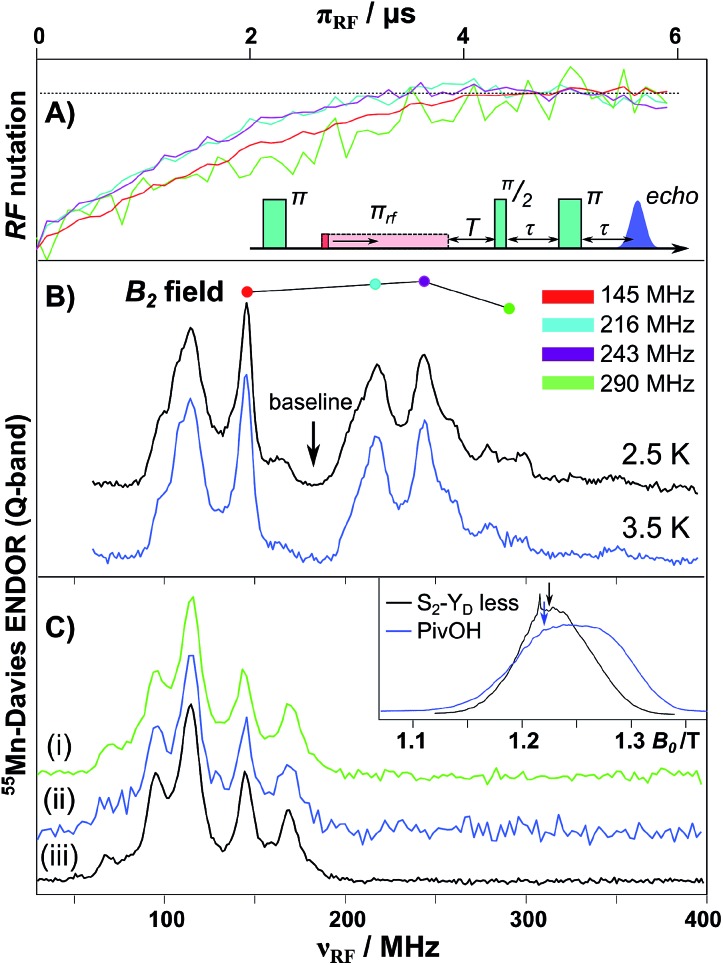
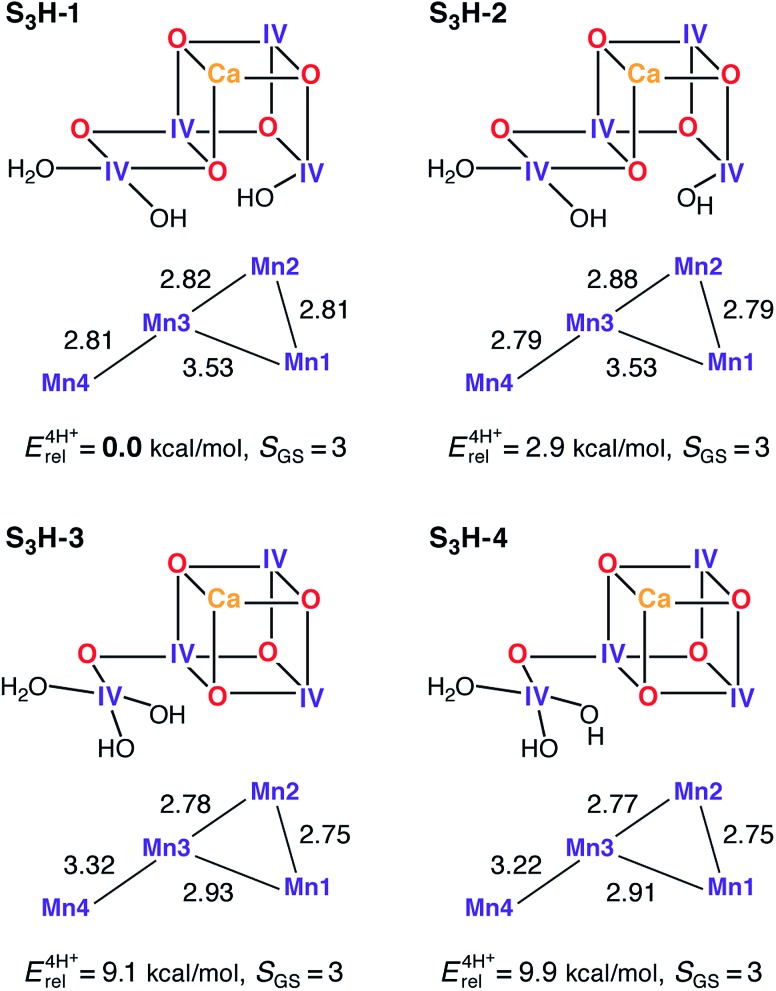
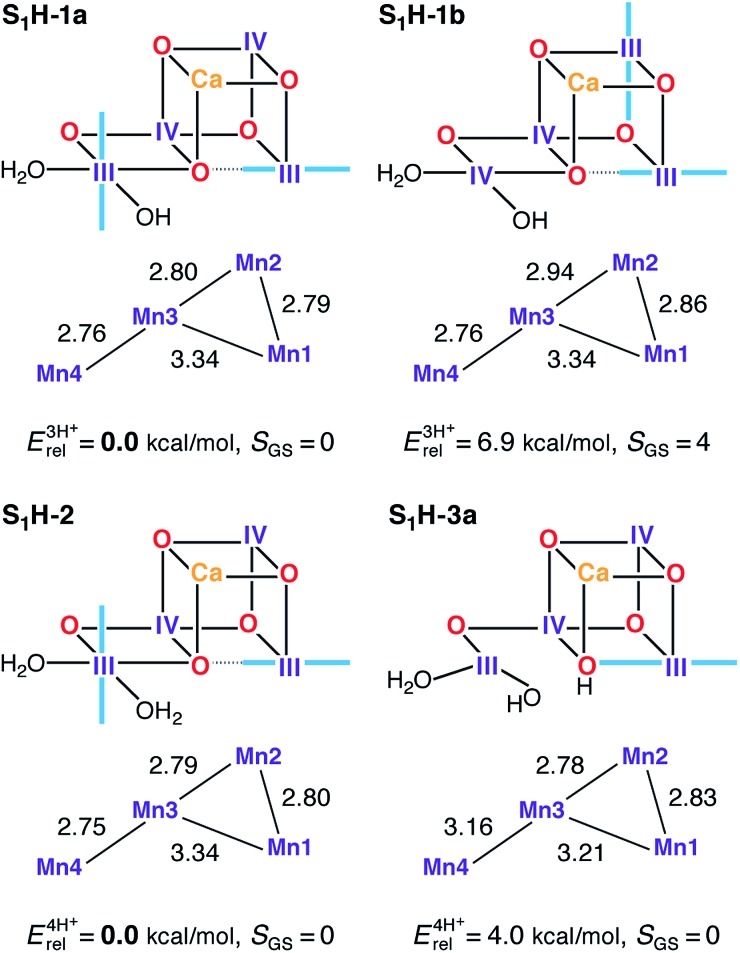
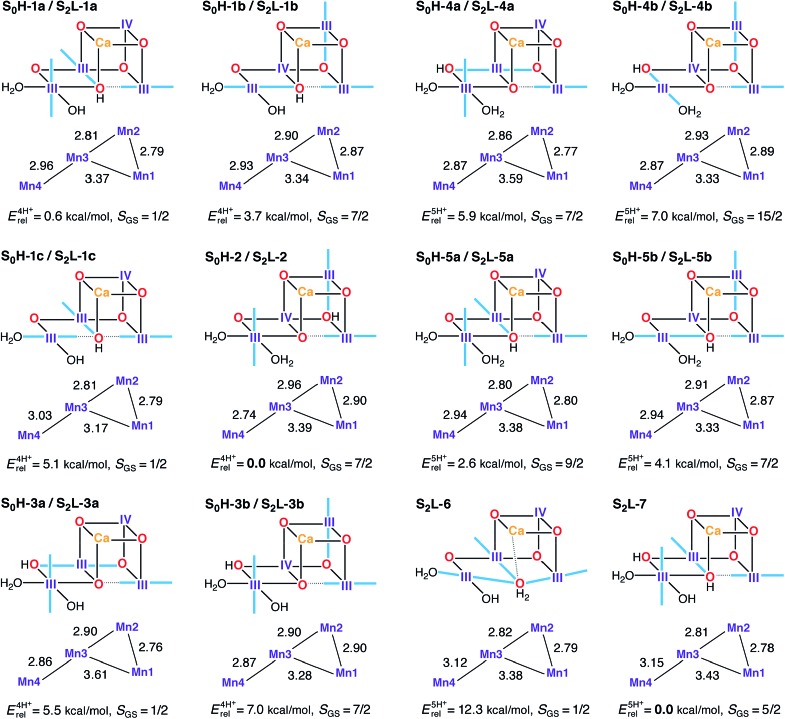
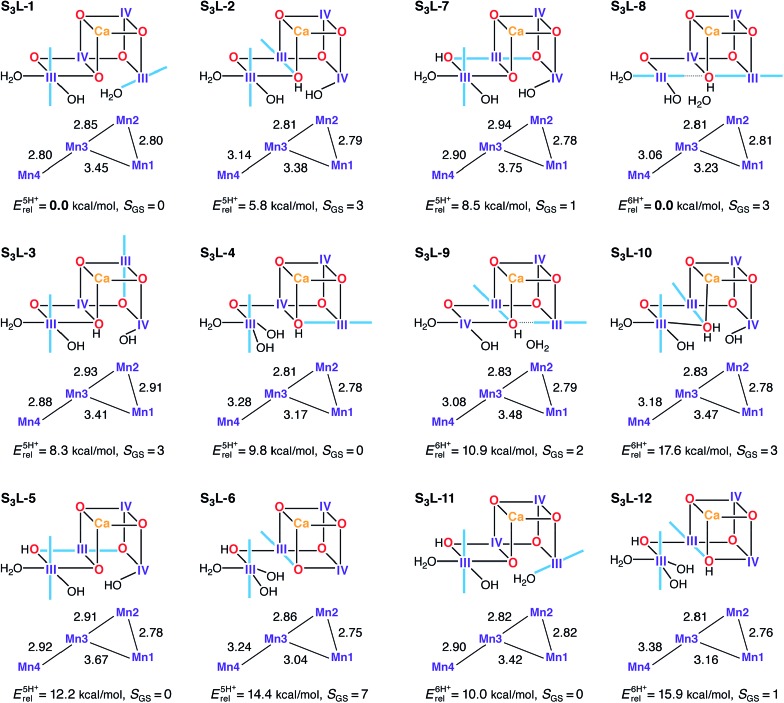
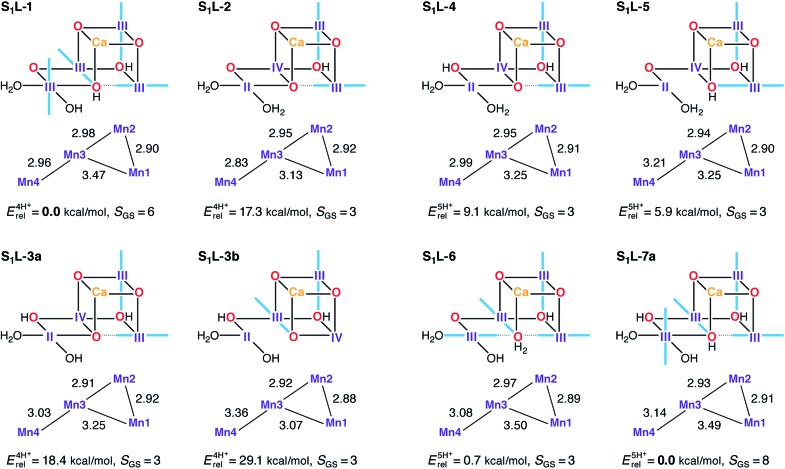
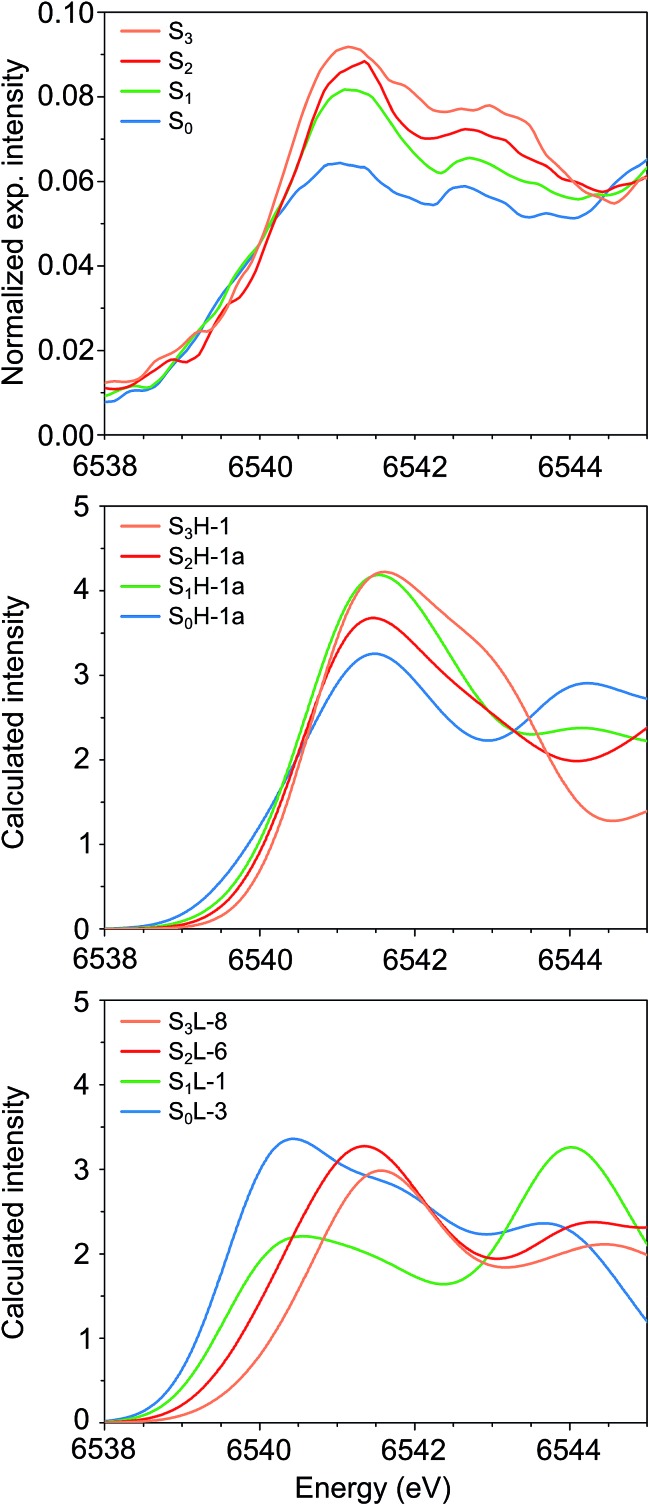
A central question in biological water splitting concerns the oxidation states of the manganese ions that comprise the oxygen-evolving complex of photosystem II. Understanding the nature and order of oxidation events that occur during the catalytic cycle of five S states ( = 0-4) is of fundamental importance both for the natural system and for artificial water oxidation catalysts. Despite the widespread adoption of the so-called "high-valent scheme"-where, for example, the Mn oxidation states in the S state are assigned as III, IV, IV, IV-the competing "low-valent scheme" that differs by a total of two metal unpaired electrons ( III, III, III, IV in the S state) is favored by several recent studies for the biological catalyst. The question of the correct oxidation state assignment is addressed here by a detailed computational comparison of the two schemes using a common structural platform and theoretical approach. Models based on crystallographic constraints were constructed for all conceivable oxidation state assignments in the four (semi)stable S states of the oxygen evolving complex, sampling various protonation levels and patterns to ensure comprehensive coverage. The models are evaluated with respect to their geometric, energetic, electronic, and spectroscopic properties against available experimental EXAFS, XFEL-XRD, EPR, ENDOR and Mn K pre-edge XANES data. New 2.5 K Mn ENDOR data of the S state are also reported. Our results conclusively show that the entire S state phenomenology can only be accommodated within the high-valent scheme by adopting a single motif and protonation pattern that progresses smoothly from S (III, III, III, IV) to S (IV, IV, IV, IV), satisfying all experimental constraints and reproducing all observables. By contrast, it was impossible to construct a consistent cycle based on the low-valent scheme for all S states. Instead, the low-valent models developed here may provide new insight into the over-reduced S states and the states involved in the assembly of the catalytically active water oxidizing cluster.
生物水分解中的一个核心问题涉及构成光系统II析氧复合物的锰离子的氧化态。了解在五个S态(S = 0 - 4)的催化循环过程中发生的氧化事件的性质和顺序,对于自然系统和人工水氧化催化剂都至关重要。尽管所谓的“高价方案”被广泛采用,例如,S态中的锰氧化态被指定为III、IV、IV、IV,但最近的几项针对生物催化剂的研究更倾向于与之竞争的“低价方案”,该方案总共相差两个金属未成对电子(S态中为III、III、III、IV)。本文通过使用共同的结构平台和理论方法对这两种方案进行详细的计算比较,来解决正确氧化态归属的问题。基于晶体学限制构建了氧释放复合物四个(半)稳定S态中所有可能的氧化态归属模型,对各种质子化水平和模式进行采样以确保全面覆盖。根据可用的实验扩展X射线吸收精细结构(EXAFS)、X射线自由电子激光 - X射线衍射(XFEL - XRD)、电子顺磁共振(EPR)、电子核双共振(ENDOR)和锰K边前X射线吸收近边结构(XANES)数据,对模型的几何、能量、电子和光谱性质进行评估。还报告了S态的新的2.5K锰ENDOR数据。我们的结果确凿地表明,只有通过采用从S(III、III、III、IV)到S(IV、IV、IV、IV)平稳进展的单一基序和质子化模式,才能在高价方案中容纳整个S态现象学,满足所有实验限制并重现所有可观测值。相比之下,基于低价方案为所有S态构建一致的循环是不可能的。相反,这里开发的低价模型可能为过度还原的S态以及参与催化活性水氧化簇组装的状态提供新的见解。