Department of Mathematics, Michigan State University, East Lansing, Michigan, United States of America.
Computer Science and Mathematics Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee, United States of America.
PLoS Comput Biol. 2018 Jan 8;14(1):e1005929. doi: 10.1371/journal.pcbi.1005929. eCollection 2018 Jan.
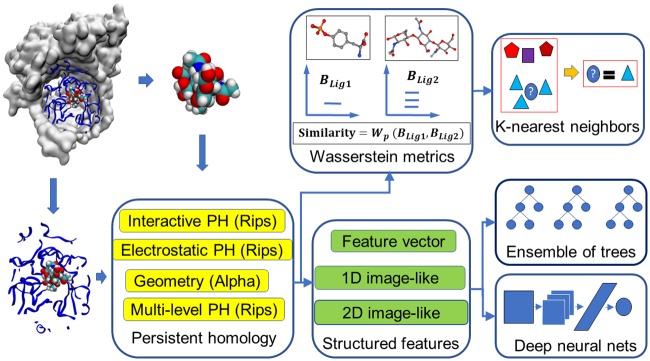
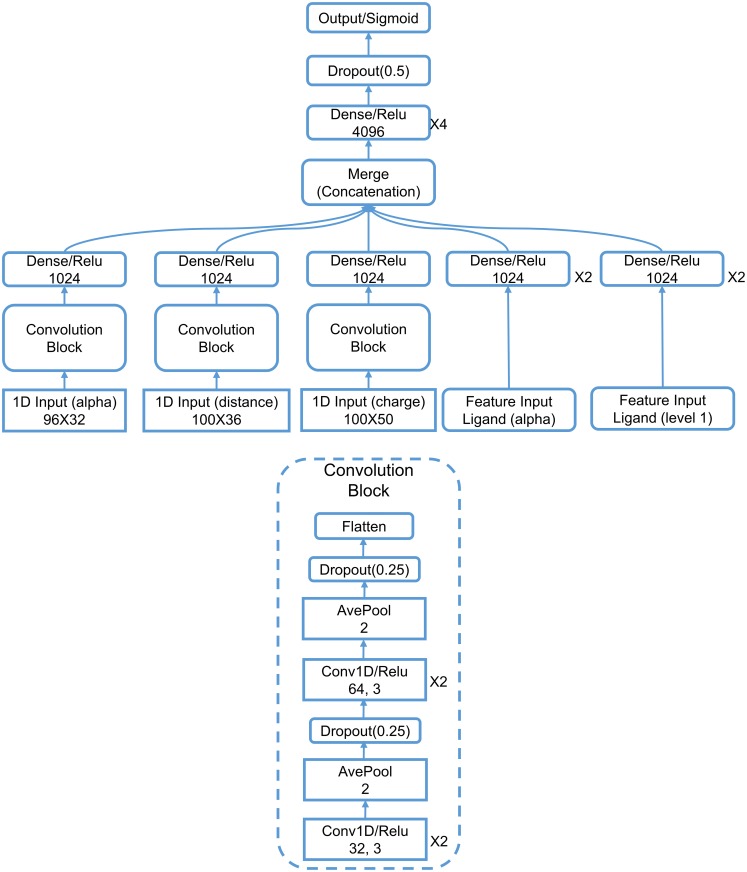
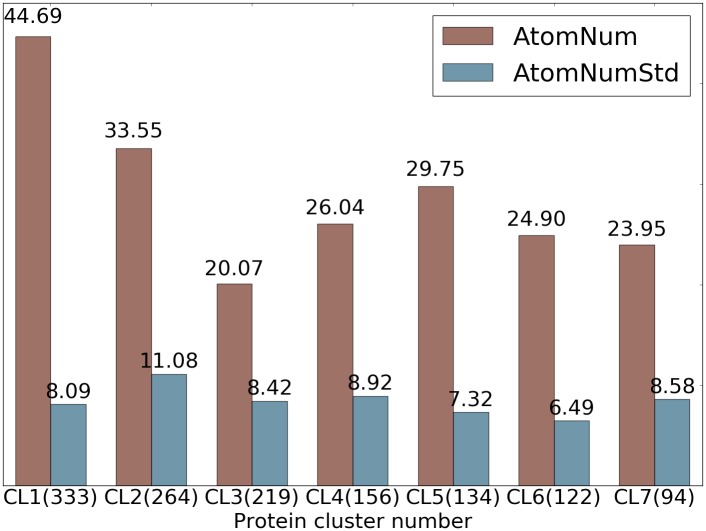
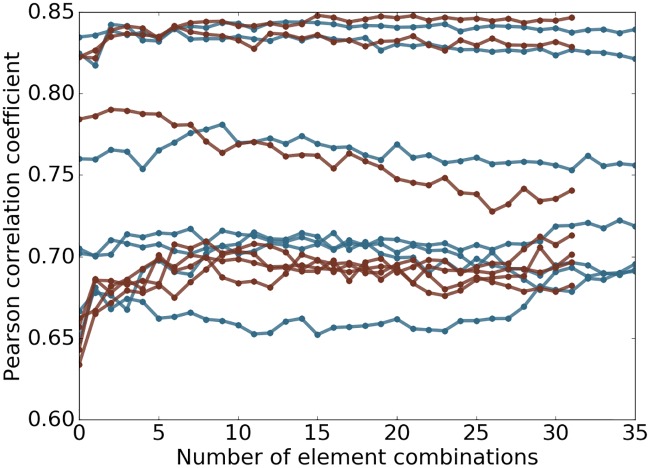
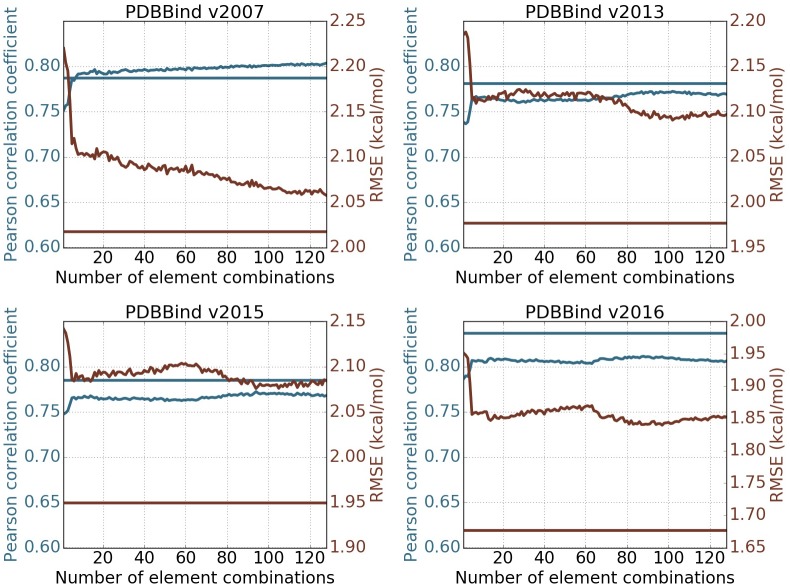
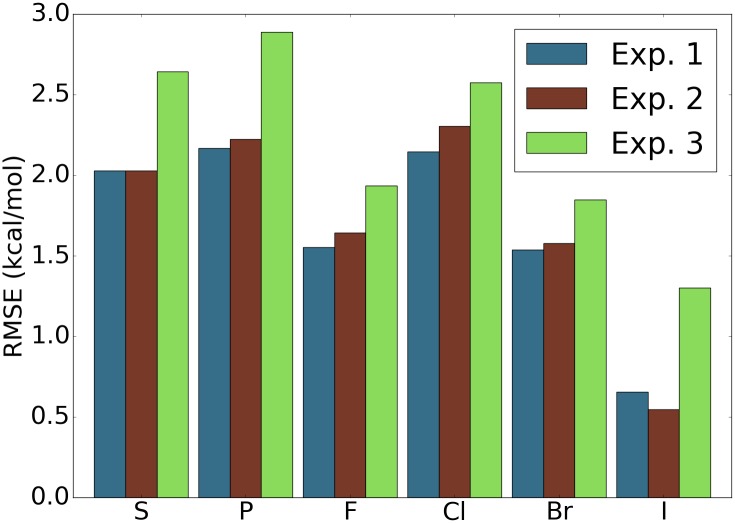
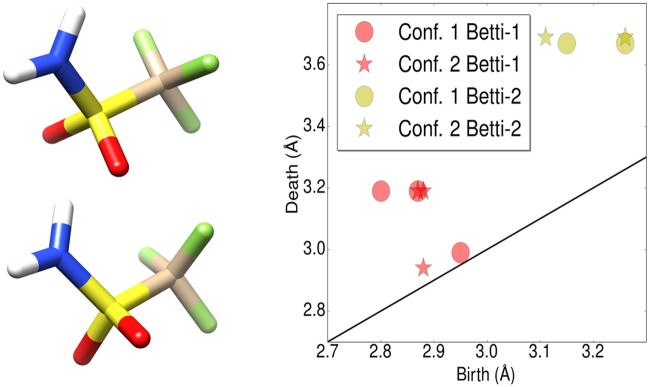
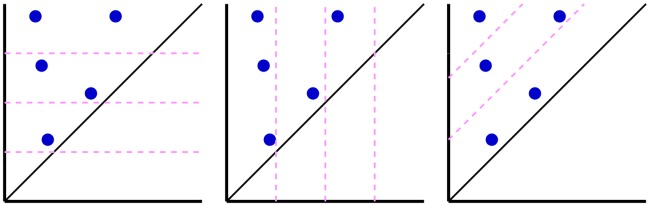
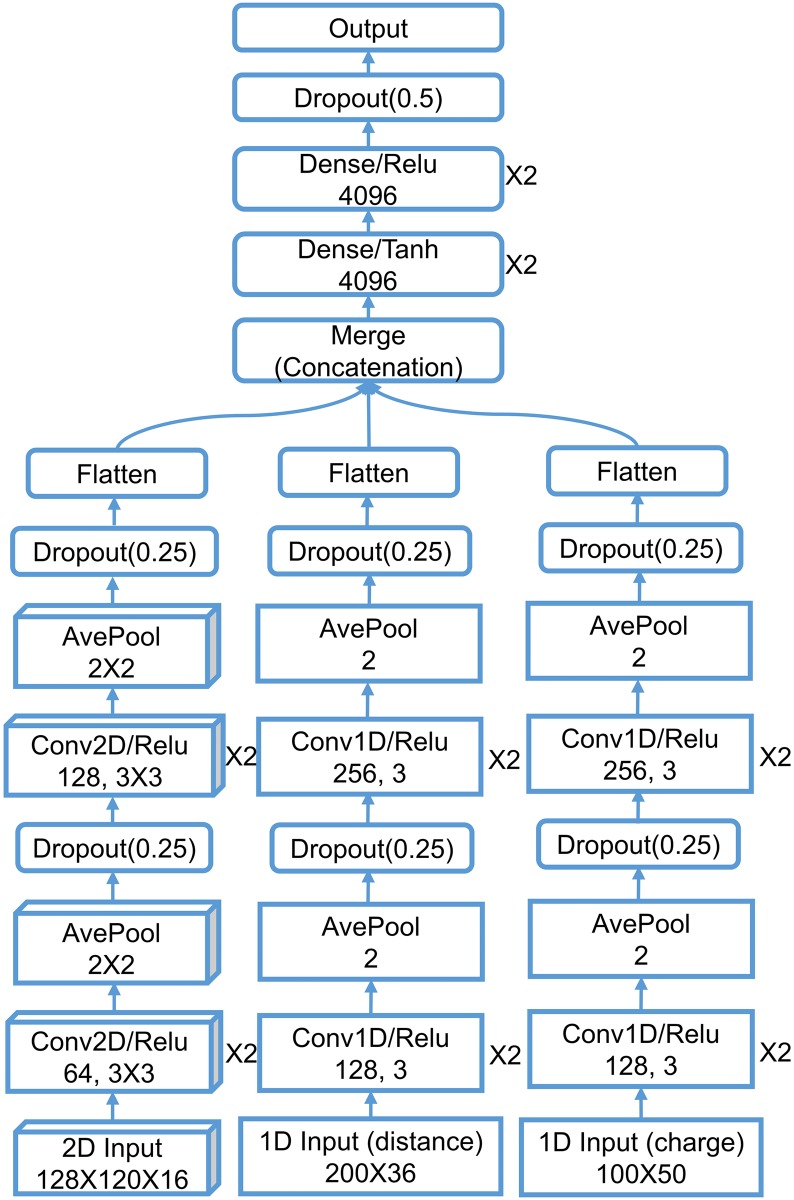
This work introduces a number of algebraic topology approaches, including multi-component persistent homology, multi-level persistent homology, and electrostatic persistence for the representation, characterization, and description of small molecules and biomolecular complexes. In contrast to the conventional persistent homology, multi-component persistent homology retains critical chemical and biological information during the topological simplification of biomolecular geometric complexity. Multi-level persistent homology enables a tailored topological description of inter- and/or intra-molecular interactions of interest. Electrostatic persistence incorporates partial charge information into topological invariants. These topological methods are paired with Wasserstein distance to characterize similarities between molecules and are further integrated with a variety of machine learning algorithms, including k-nearest neighbors, ensemble of trees, and deep convolutional neural networks, to manifest their descriptive and predictive powers for protein-ligand binding analysis and virtual screening of small molecules. Extensive numerical experiments involving 4,414 protein-ligand complexes from the PDBBind database and 128,374 ligand-target and decoy-target pairs in the DUD database are performed to test respectively the scoring power and the discriminatory power of the proposed topological learning strategies. It is demonstrated that the present topological learning outperforms other existing methods in protein-ligand binding affinity prediction and ligand-decoy discrimination.
这项工作介绍了一些代数拓扑方法,包括多分量持久同调、多层次持久同调和静电持久,用于小分子和生物分子复合物的表示、特征描述和描述。与传统的持久同调相比,多分量持久同调在生物分子几何复杂性的拓扑简化过程中保留了关键的化学和生物学信息。多层次持久同调能够对感兴趣的分子内和/或分子间相互作用进行定制化的拓扑描述。静电持久将部分电荷信息纳入拓扑不变量中。这些拓扑方法与 Wasserstein 距离相结合,用于表征分子之间的相似性,并进一步与各种机器学习算法(包括 k-最近邻、树集成和深度卷积神经网络)集成,以展示它们在蛋白质-配体结合分析和小分子虚拟筛选中的描述和预测能力。进行了广泛的数值实验,涉及来自 PDBBind 数据库的 4414 个蛋白质-配体复合物和来自 DUD 数据库的 128374 个配体-靶标和诱饵-靶标对,分别测试了所提出的拓扑学习策略的评分能力和区分能力。结果表明,与其他现有方法相比,本拓扑学习方法在蛋白质-配体结合亲和力预测和配体-诱饵区分方面表现更好。