Arruda Maria Augusta, Stoddart Leigh A, Gherbi Karolina, Briddon Stephen J, Kellam Barrie, Hill Stephen J
Division of Physiology, Pharmacology and Neuroscience, School of Life Sciences, Medical School, Queen's Medical Centre, University of Nottingham, Nottingham, United Kingdom.
Centre of Membrane Proteins and Receptors, University of Birmingham and University of Nottingham, The Midlands, United Kingdom.
Front Pharmacol. 2017 Dec 13;8:908. doi: 10.3389/fphar.2017.00908. eCollection 2017.
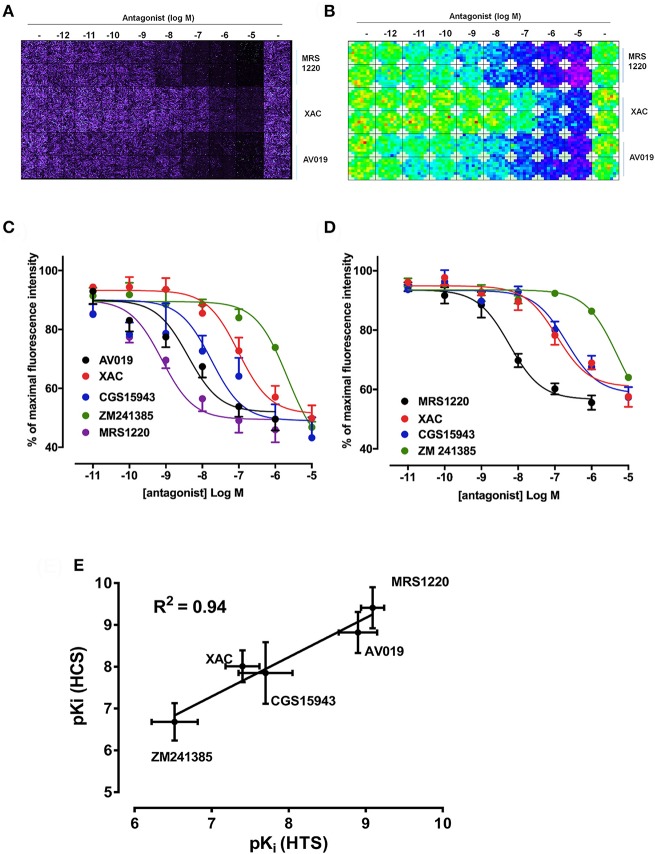
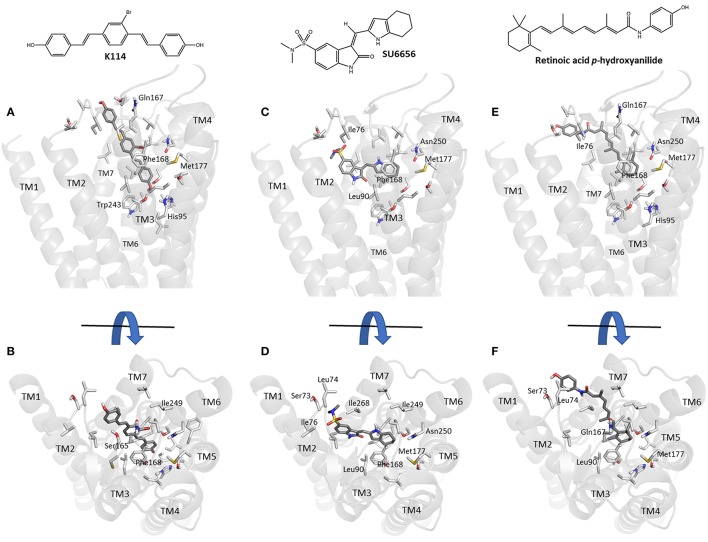
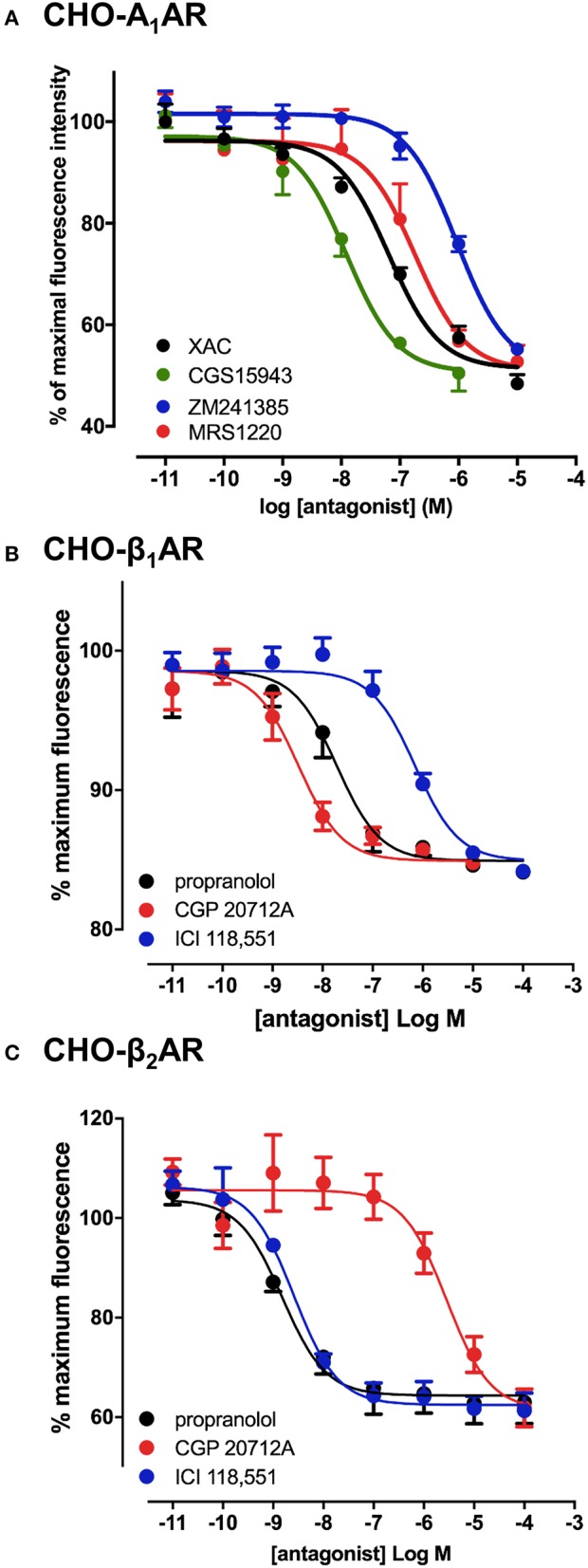
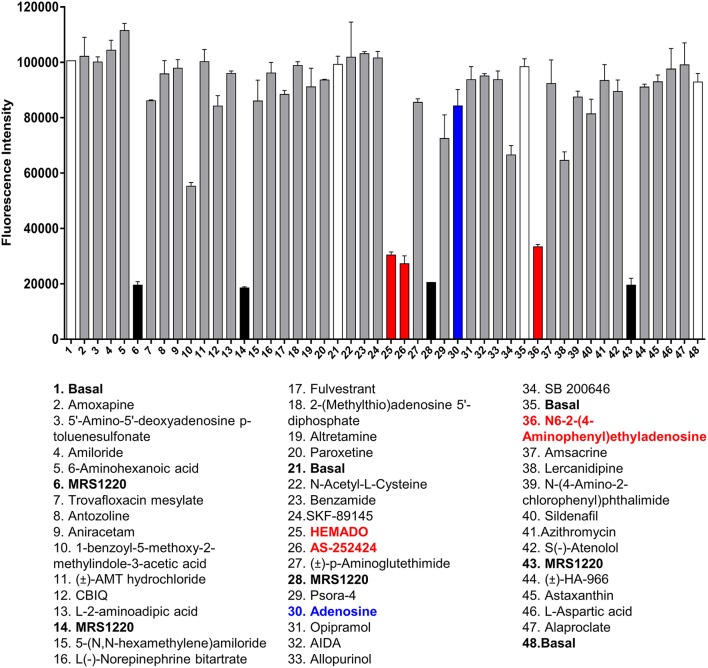
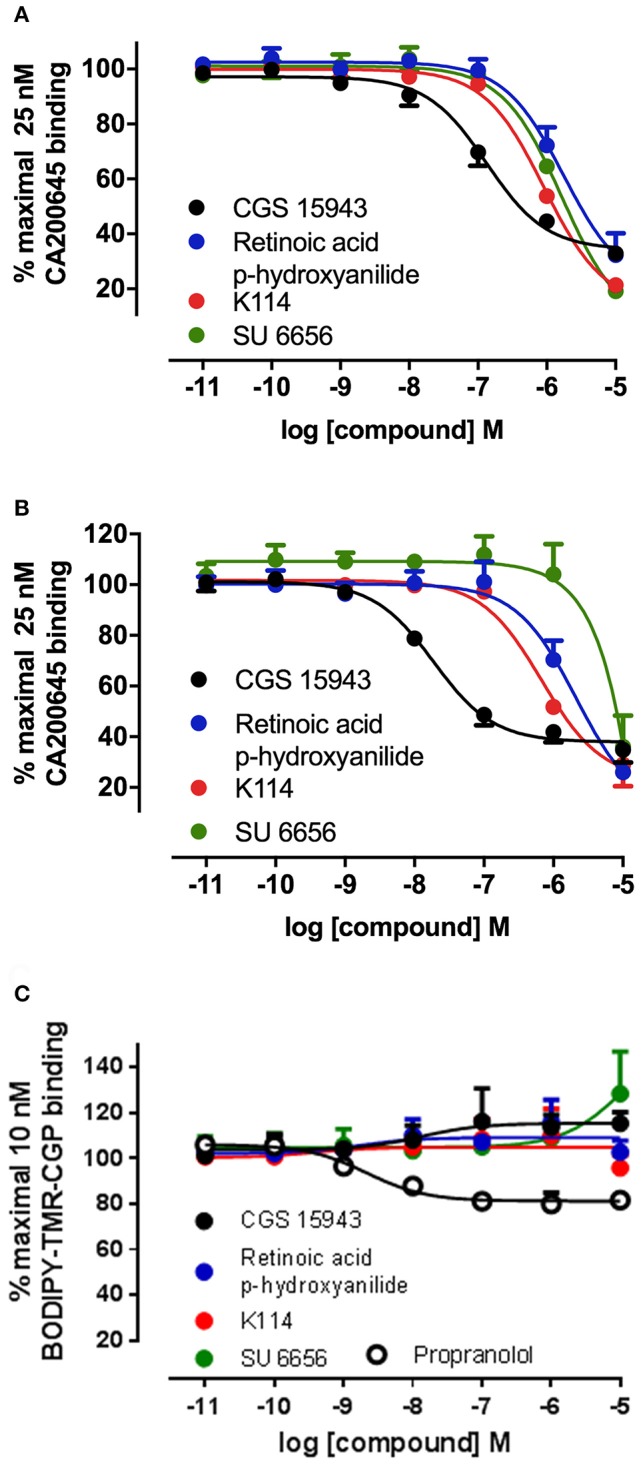
Recent advances in fluorescent ligand technology have enabled the study of G protein-coupled receptors in their native environment without the need for genetic modification such as addition of N-terminal fluorescent or bioluminescent tags. Here, we have used a non-imaging plate reader (PHERAstar FS) to monitor the binding of fluorescent ligands to the human adenosine-A receptor (AAR; CA200645 and AV039), stably expressed in CHO-K1 cells. To verify that this method was suitable for the study of other GPCRs, assays at the human adenosine-A receptor, and β and β adrenoceptors (βAR and βAR; BODIPY-TMR-CGP-12177) were also carried out. Affinity values determined for the binding of the fluorescent ligands CA200645 and AV039 to AAR for a range of classical adenosine receptor antagonists were consistent with AAR pharmacology and correlated well ( = 0.94) with equivalent data obtained using a confocal imaging plate reader (ImageXpress Ultra). The binding of BODIPY-TMR-CGP-12177 to the βAR was potently inhibited by low concentrations of the β-selective antagonist CGP 20712A (pK 9.68) but not by the β-selective antagonist ICI 118551(pK 7.40). Furthermore, in experiments conducted in CHO K1 cells expressing the βAR this affinity order was reversed with ICI 118551 showing the highest affinity (pK 8.73) and CGP20712A (pK 5.68) the lowest affinity. To determine whether the faster data acquisition of the non-imaging plate reader (~3 min per 96-well plate) was suitable for high throughput screening (HTS), we screened the LOPAC library for inhibitors of the binding of CA200645 to the AAR. From the initial 1,263 compounds evaluated, 67 hits (defined as those that inhibited the total binding of 25 nM CA200645 by ≥40%) were identified. All compounds within the library that had medium to high affinity for the AAR (pK ≥6) were successfully identified. We found three novel compounds in the library that displayed unexpected sub-micromolar affinity for the AAR. These were K114 (pK 6.43), retinoic acid -hydroxyanilide (pK 6.13) and SU 6556 (pK 6.17). Molecular docking of these latter three LOPAC library members provided a plausible set of binding poses within the vicinity of the established orthosteric AAR binding pocket. A plate reader based library screening using an untagged receptor is therefore possible using fluorescent ligand opening the possibility of its use in compound screening at natively expressed receptors.
荧光配体技术的最新进展使得在天然环境中研究G蛋白偶联受体成为可能,而无需进行基因改造,如添加N端荧光或生物发光标签。在此,我们使用非成像酶标仪(PHERAstar FS)监测荧光配体与稳定表达于CHO-K1细胞中的人腺苷A受体(AAR;CA200645和AV039)的结合。为验证该方法是否适用于其他GPCR的研究,我们还对人腺苷A受体以及β1和β2肾上腺素能受体(β1AR和β2AR;BODIPY-TMR-CGP-12177)进行了检测。测定了一系列经典腺苷受体拮抗剂存在时荧光配体CA200645和AV039与AAR结合的亲和力值,这些值与AAR药理学一致,并且与使用共聚焦成像酶标仪(ImageXpress Ultra)获得的等效数据具有良好的相关性(r = 0.94)。低浓度的β1选择性拮抗剂CGP 20712A(pK 9.68)可有效抑制BODIPY-TMR-CGP-12177与β1AR的结合,而β2选择性拮抗剂ICI 118551(pK 7.40)则无此作用。此外,在表达β2AR的CHO K1细胞中进行的实验中,这种亲和力顺序发生了逆转,ICI 118551显示出最高亲和力(pK 8.73),而CGP20712A(pK 5.68)显示出最低亲和力。为确定非成像酶标仪更快的数据采集速度(每96孔板约3分钟)是否适用于高通量筛选(HTS),我们筛选了LOPAC文库中CA200645与AAR结合的抑制剂。从最初评估的1263种化合物中,鉴定出67个活性命中物(定义为那些抑制25 nM CA200645总结合≥40%的化合物)。文库中所有对AAR具有中等到高亲和力(pK≥6)的化合物均被成功鉴定。我们在文库中发现了三种对AAR显示出意外的亚微摩尔亲和力的新型化合物。它们是K114(pK 6.43)、维甲酸-4-羟基苯胺(pK 6.13)和SU 6556(pK 6.17)。对后三种LOPAC文库成员进行分子对接,在已确定的正构AAR结合口袋附近提供了一组合理的结合构象。因此,使用荧光配体基于酶标仪的文库筛选在未标记受体的情况下是可行的,这为其在天然表达受体的化合物筛选中的应用开辟了可能性。