Fuwai Hospital, State Key Laboratory of Cardiovascular Diseases, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100037, P.R. China.
Novogene Bioinformatics Institute, Beijing, 100000, P.R. China.
Sci Rep. 2018 Jan 12;8(1):635. doi: 10.1038/s41598-017-18756-2.
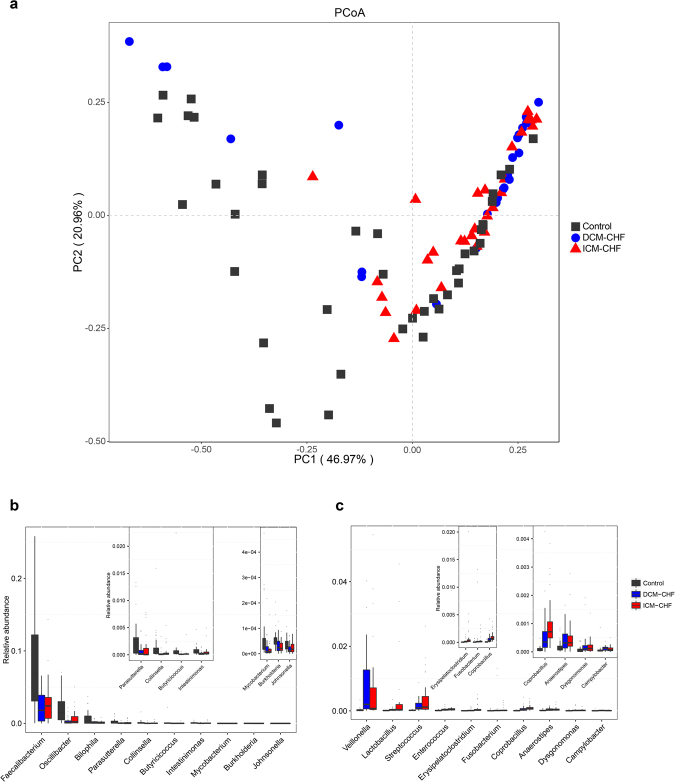
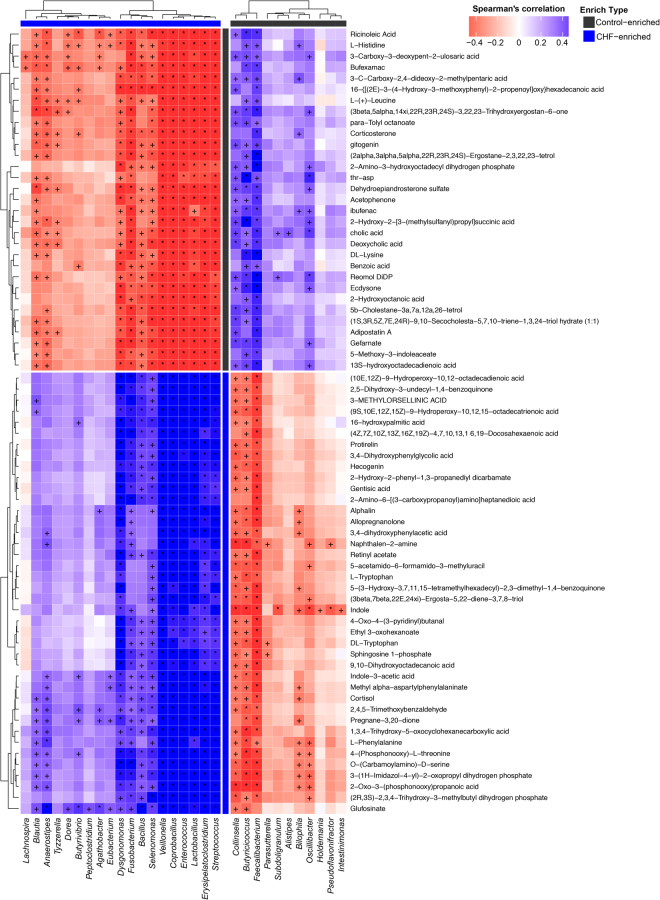
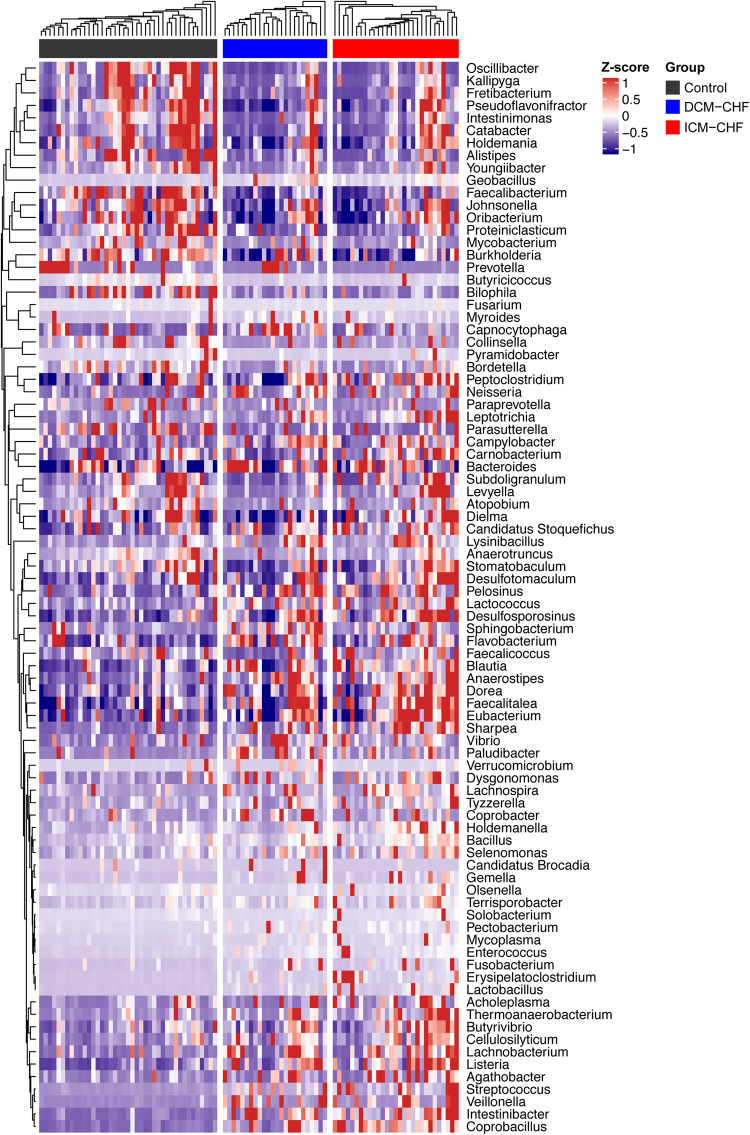
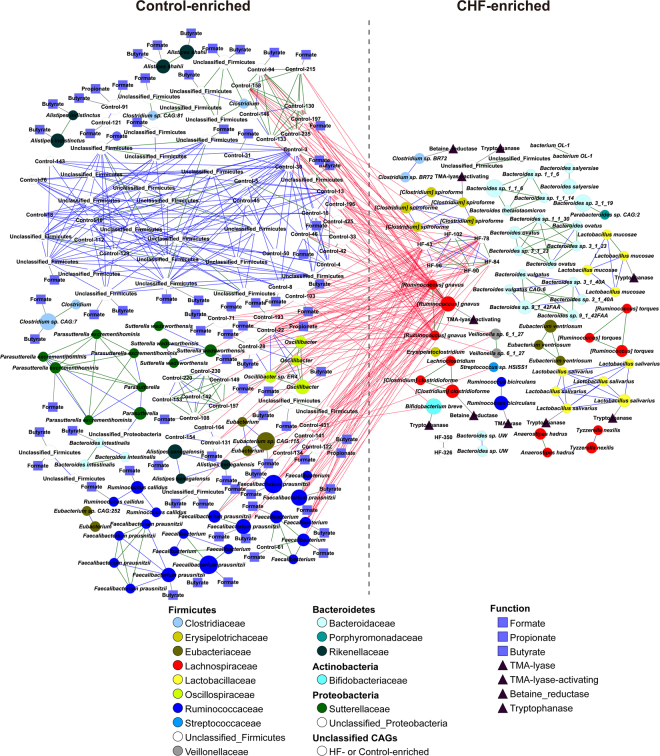
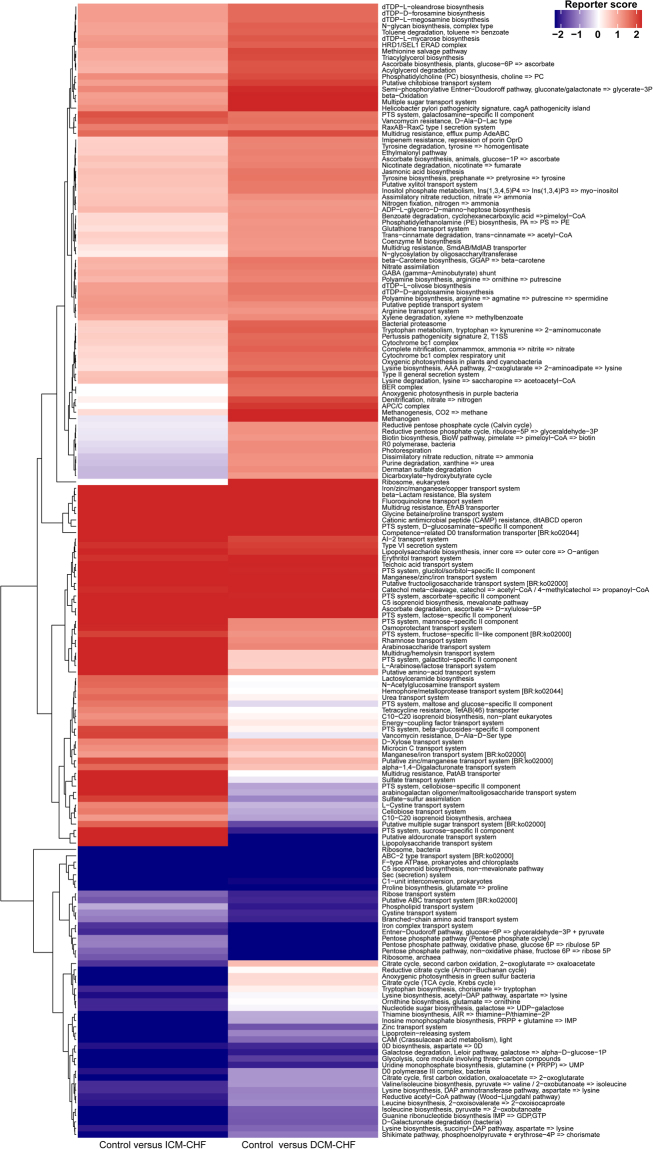
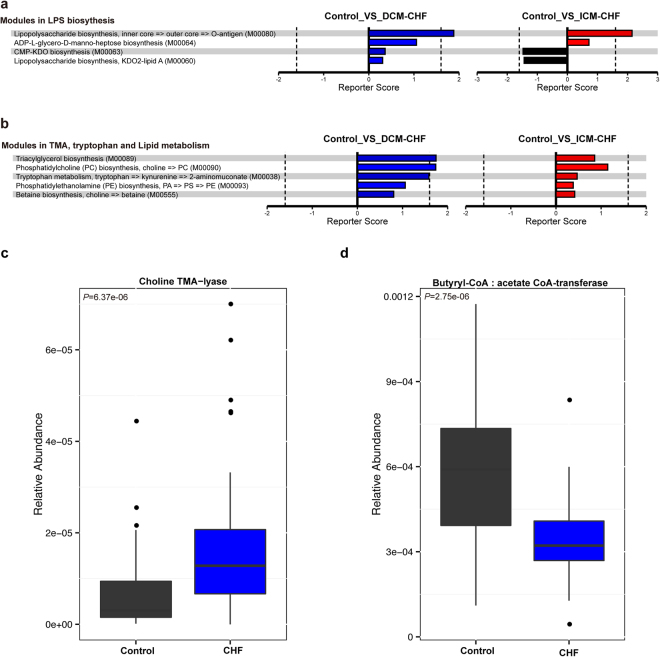
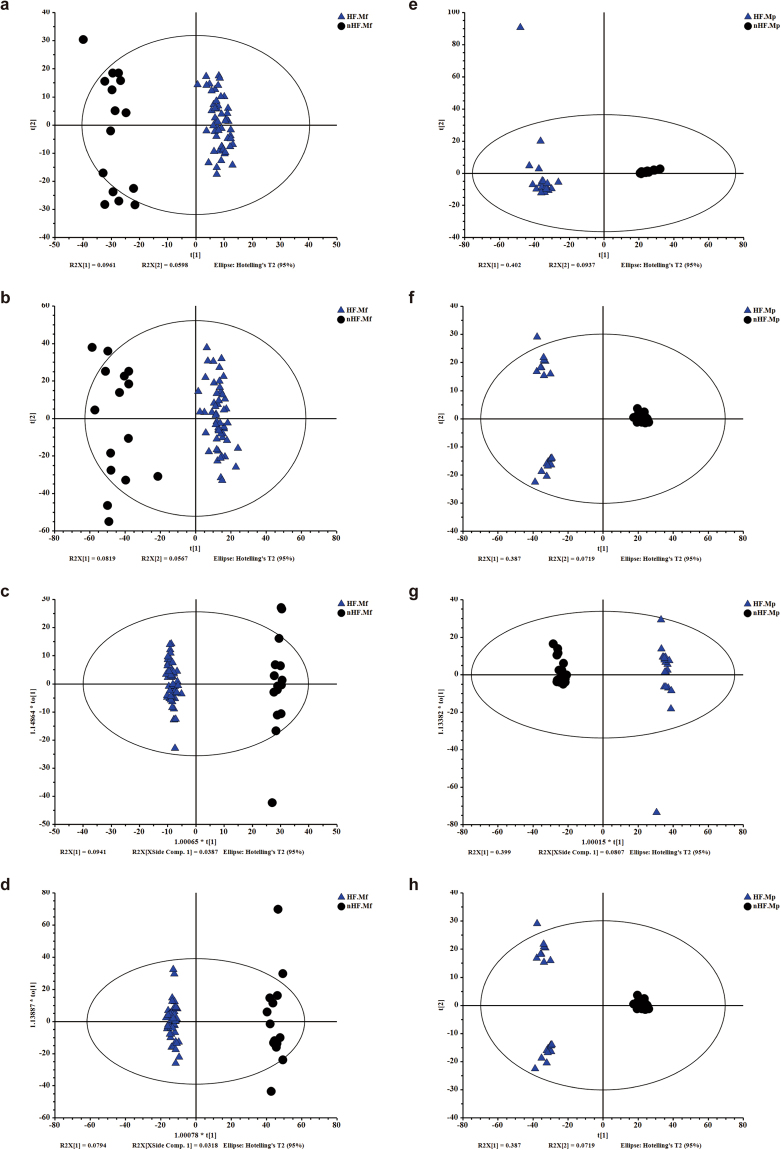
Previous studies suggested a possible gut microbiota dysbiosis in chronic heart failure (CHF). However, direct evidence was lacking. In this study, we investigated the composition and metabolic patterns of gut microbiota in CHF patients to provide direct evidence and comprehensive understanding of gut microbiota dysbiosis in CHF. We enrolled 53 CHF patients and 41 controls. Metagenomic analyses of faecal samples and metabolomic analyses of faecal and plasma samples were then performed. We found that the composition of gut microbiota in CHF was significantly different from controls. Faecalibacterium prausnitzii decrease and Ruminococcus gnavus increase were the essential characteristics in CHF patients' gut microbiota. We also observed an imbalance of gut microbes involved in the metabolism of protective metabolites such as butyrate and harmful metabolites such as trimethylamine N-oxide in CHF patients. Metabolic features of both faecal and plasma samples from CHF patients also significantly changed. Moreover, alterations in faecal and plasma metabolic patterns correlated with gut microbiota dysbiosis in CHF. Taken together, we found that CHF was associated with distinct gut microbiota dysbiosis and pinpointed the specific core bacteria imbalance in CHF, along with correlations between changes in certain metabolites and gut microbes.
先前的研究表明,慢性心力衰竭(CHF)患者可能存在肠道微生物群落失调。然而,这方面直接证据的缺乏。在这项研究中,我们调查了 CHF 患者肠道微生物群落的组成和代谢模式,为 CHF 患者肠道微生物群落失调提供了直接证据和全面的认识。我们纳入了 53 名 CHF 患者和 41 名对照者。然后对粪便样本进行宏基因组学分析,对粪便和血浆样本进行代谢组学分析。我们发现 CHF 患者肠道微生物群落的组成与对照者有显著差异。Faecalibacterium prausnitzii 的减少和 Ruminococcus gnavus 的增加是 CHF 患者肠道微生物群落的主要特征。我们还观察到 CHF 患者涉及保护性代谢物(如丁酸)和有害代谢物(如三甲胺 N-氧化物)代谢的肠道微生物失衡。CHF 患者粪便和血浆样本的代谢特征也明显改变。此外,粪便和血浆代谢模式的改变与 CHF 患者肠道微生物群落失调相关。总之,我们发现 CHF 与明显的肠道微生物群落失调有关,并确定了 CHF 中特定的核心细菌失衡,以及某些代谢物和肠道微生物之间的相关性。