Department of Stem Cell Biology and Regenerative Medicine, Keck School of Medicine, University of Southern California, Los Angeles, California, USA.
Eli and Edythe Broad CIRM Center for Regenerative Medicine and Stem Cell Research at USC, Los Angeles, California, USA.
Nat Med. 2018 Mar;24(3):313-325. doi: 10.1038/nm.4490. Epub 2018 Feb 5.
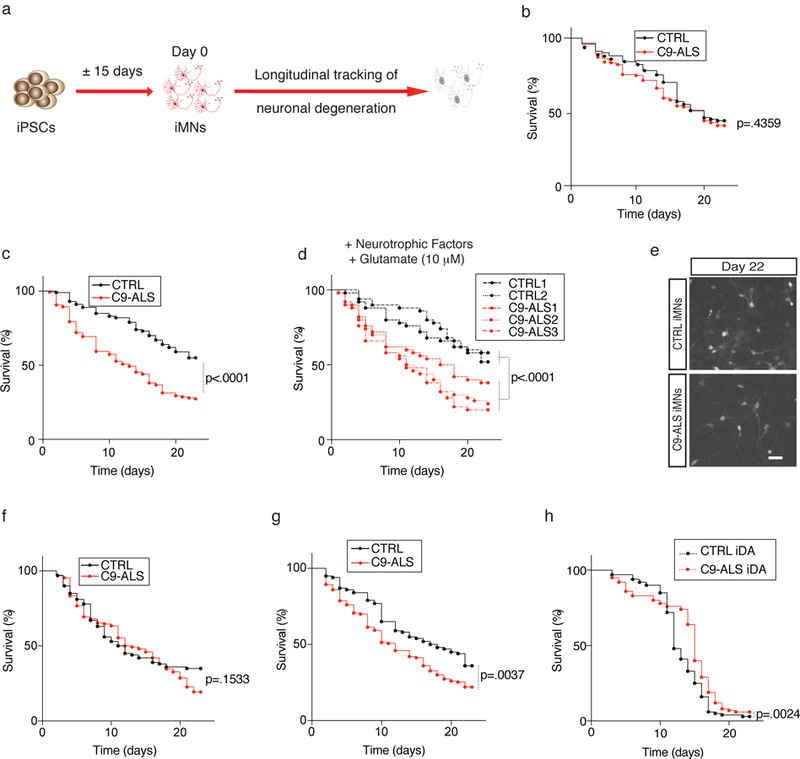
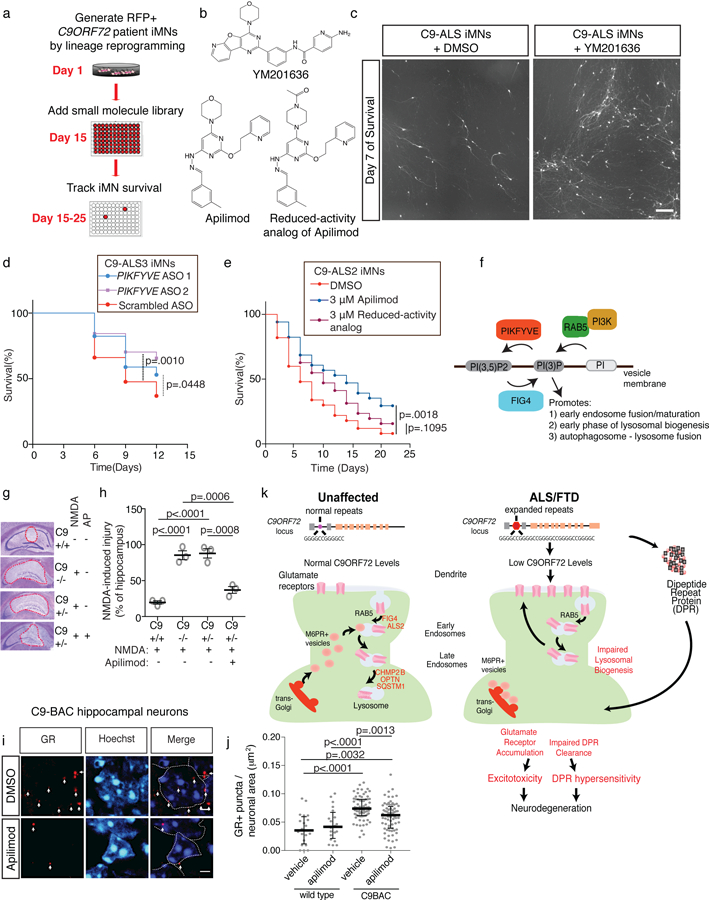
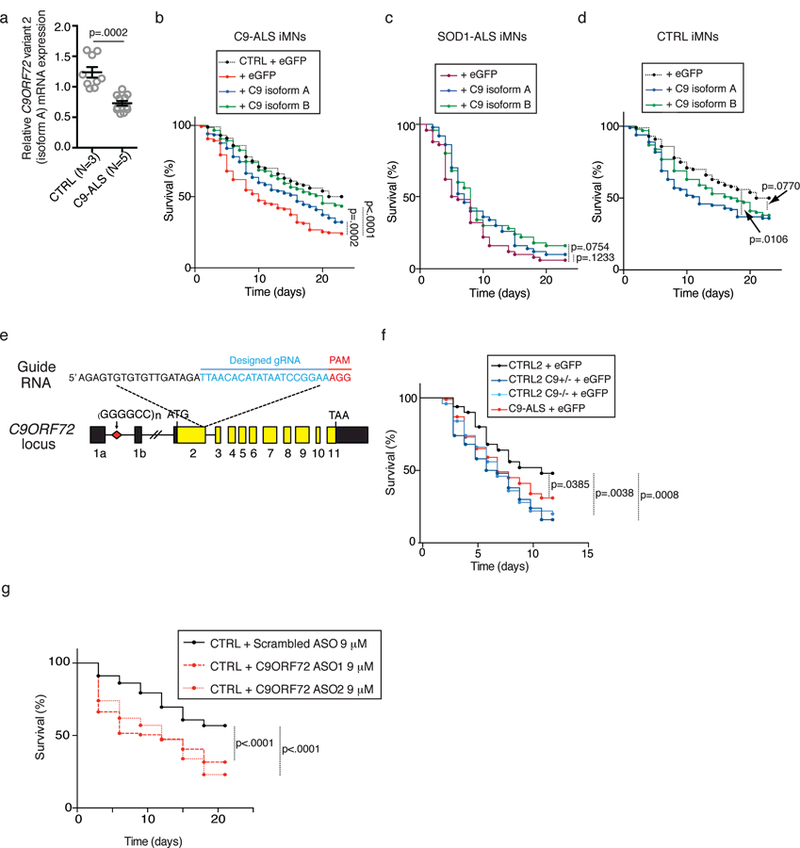
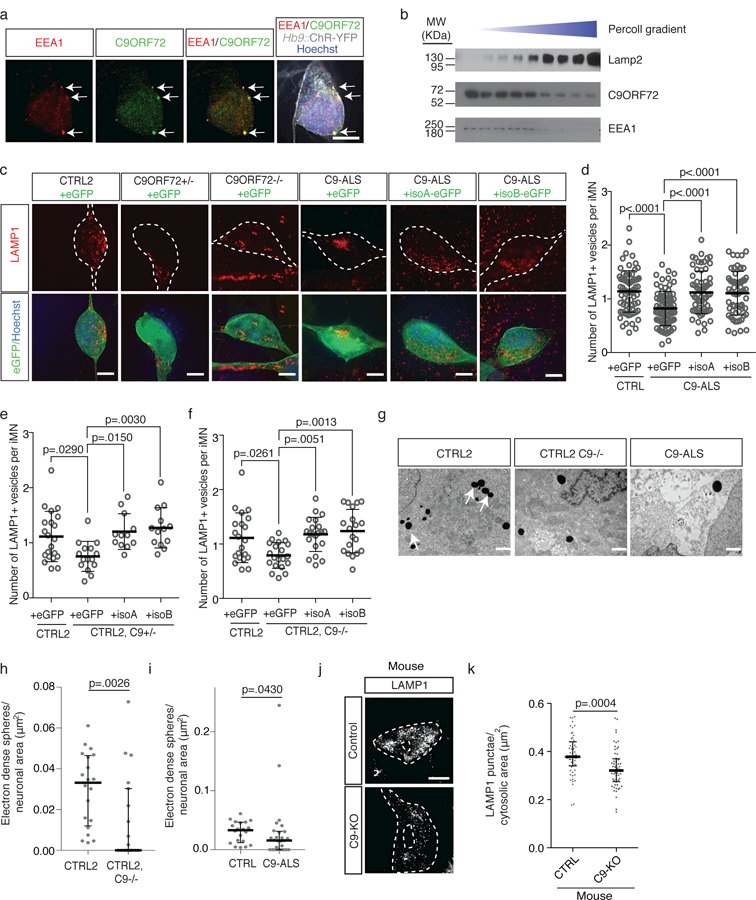
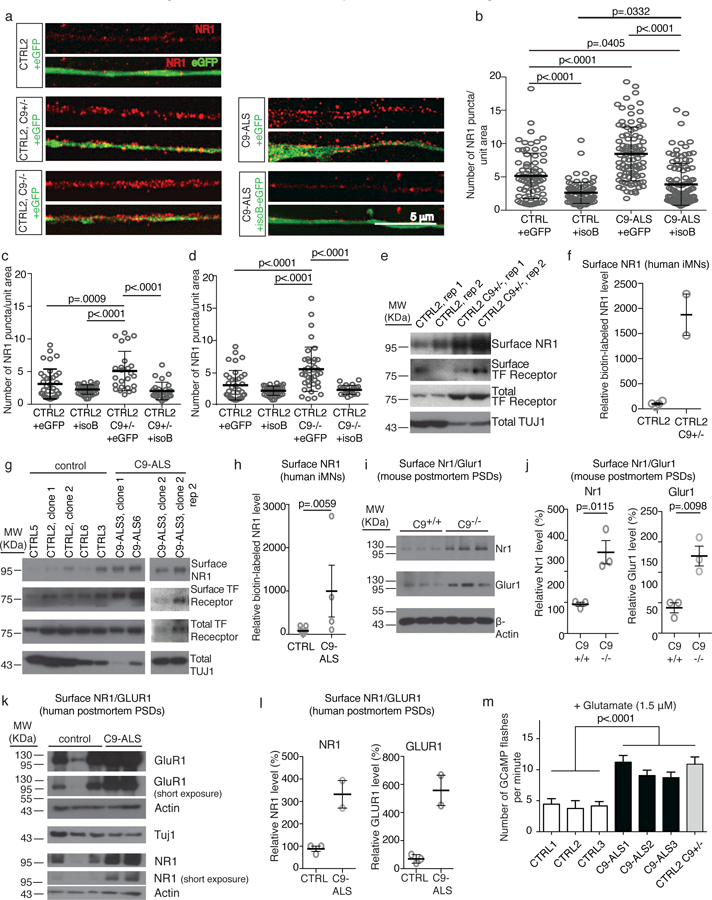
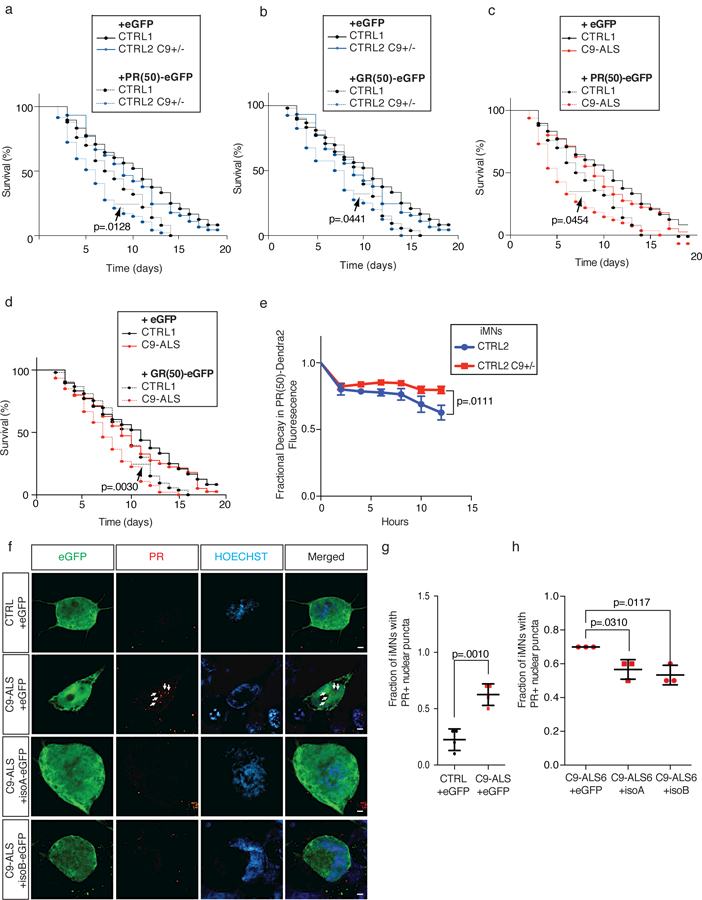
An intronic GGGGCC repeat expansion in C9ORF72 is the most common cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), but the pathogenic mechanism of this repeat remains unclear. Using human induced motor neurons (iMNs), we found that repeat-expanded C9ORF72 was haploinsufficient in ALS. We found that C9ORF72 interacted with endosomes and was required for normal vesicle trafficking and lysosomal biogenesis in motor neurons. Repeat expansion reduced C9ORF72 expression, triggering neurodegeneration through two mechanisms: accumulation of glutamate receptors, leading to excitotoxicity, and impaired clearance of neurotoxic dipeptide repeat proteins derived from the repeat expansion. Thus, cooperativity between gain- and loss-of-function mechanisms led to neurodegeneration. Restoring C9ORF72 levels or augmenting its function with constitutively active RAB5 or chemical modulators of RAB5 effectors rescued patient neuron survival and ameliorated neurodegenerative processes in both gain- and loss-of-function C9ORF72 mouse models. Thus, modulating vesicle trafficking was able to rescue neurodegeneration caused by the C9ORF72 repeat expansion. Coupled with rare mutations in ALS2, FIG4, CHMP2B, OPTN and SQSTM1, our results reveal mechanistic convergence on vesicle trafficking in ALS and FTD.
C9ORF72 基因中的内含子 GGGGCC 重复扩展是肌萎缩侧索硬化症(ALS)和额颞叶痴呆(FTD)最常见的原因,但这种重复的致病机制仍不清楚。我们使用人类诱导的运动神经元(iMNs)发现,重复扩展的 C9ORF72 在 ALS 中表现为单倍不足。我们发现 C9ORF72 与内体相互作用,是运动神经元中正常囊泡运输和溶酶体生物发生所必需的。重复扩展降低了 C9ORF72 的表达,通过两种机制引发神经退行性变:谷氨酸受体的积累,导致兴奋性毒性,以及重复扩展衍生的神经毒性二肽重复蛋白的清除受损。因此,功能获得和功能丧失机制之间的协同作用导致了神经退行性变。恢复 C9ORF72 水平或通过组成型激活 RAB5 或 RAB5 效应物的化学调节剂增强其功能,挽救了患者神经元的存活,并改善了 gain- 和 loss-of-function C9ORF72 小鼠模型中的神经退行性过程。因此,调节囊泡运输能够挽救由 C9ORF72 重复扩展引起的神经退行性变。结合 ALS2、FIG4、CHMP2B、OPTN 和 SQSTM1 中的罕见突变,我们的结果揭示了 ALS 和 FTD 中囊泡运输的机制收敛。