Department of Gastroenterology, The University of Tokyo, Tokyo, Japan.
Division of Gastroenterology, Institute for Adult Diseases, Asahi Life Foundation, Tokyo, Japan.
Gut. 2018 Aug;67(8):1493-1504. doi: 10.1136/gutjnl-2017-315193. Epub 2018 Feb 6.
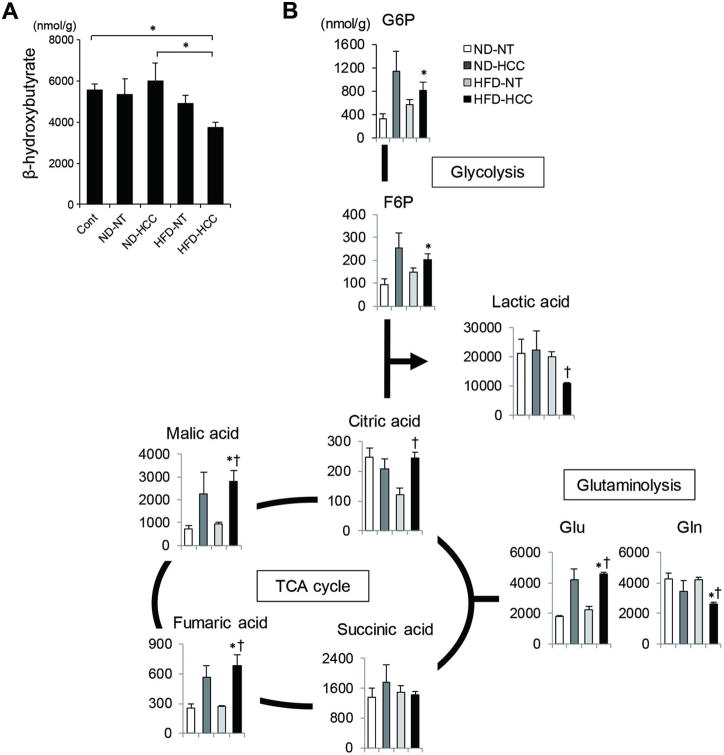
Metabolic reprogramming of tumour cells that allows for adaptation to their local environment is a hallmark of cancer. Interestingly, obesity-driven and non-alcoholic steatohepatitis (NASH)-driven hepatocellular carcinoma (HCC) mouse models commonly exhibit strong steatosis in tumour cells as seen in human steatohepatitic HCC (SH-HCC), which may reflect a characteristic metabolic alteration.
Non-tumour and HCC tissues obtained from diethylnitrosamine-injected mice fed either a normal or a high-fat diet (HFD) were subjected to comprehensive metabolome analysis, and the significance of obesity-mediated metabolic alteration in hepatocarcinogenesis was evaluated.
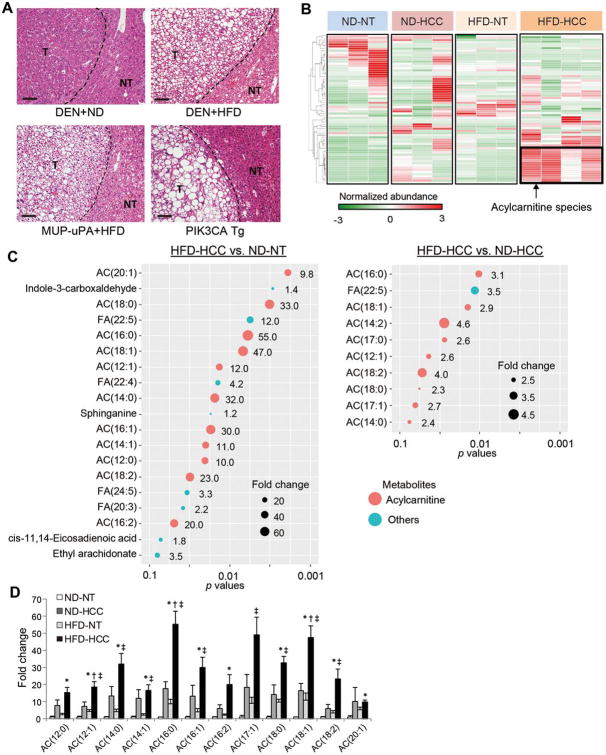
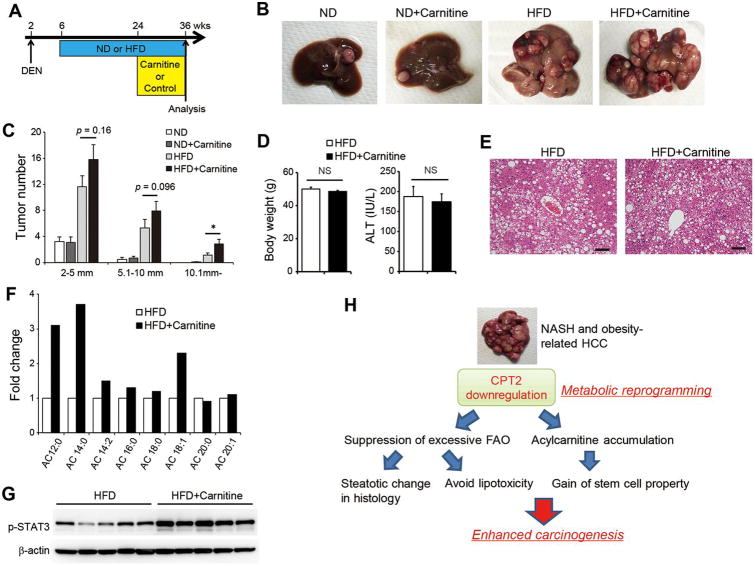
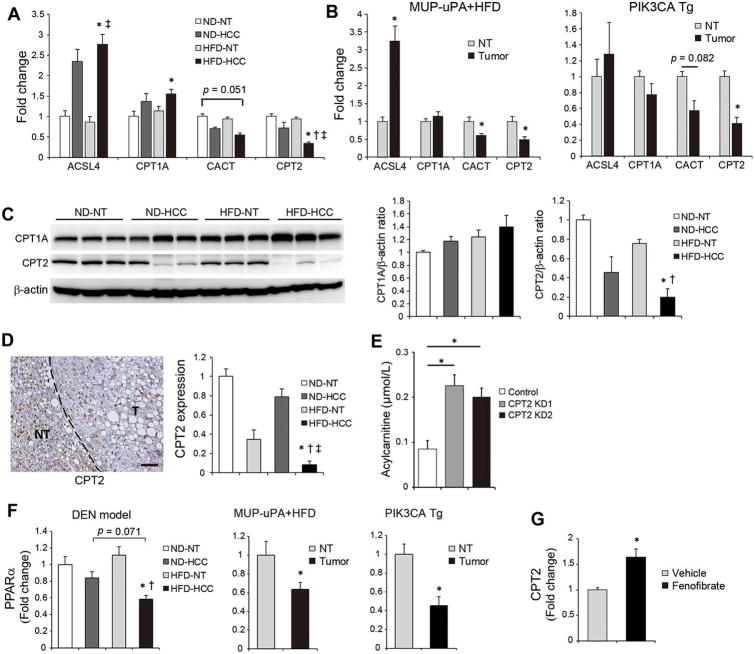
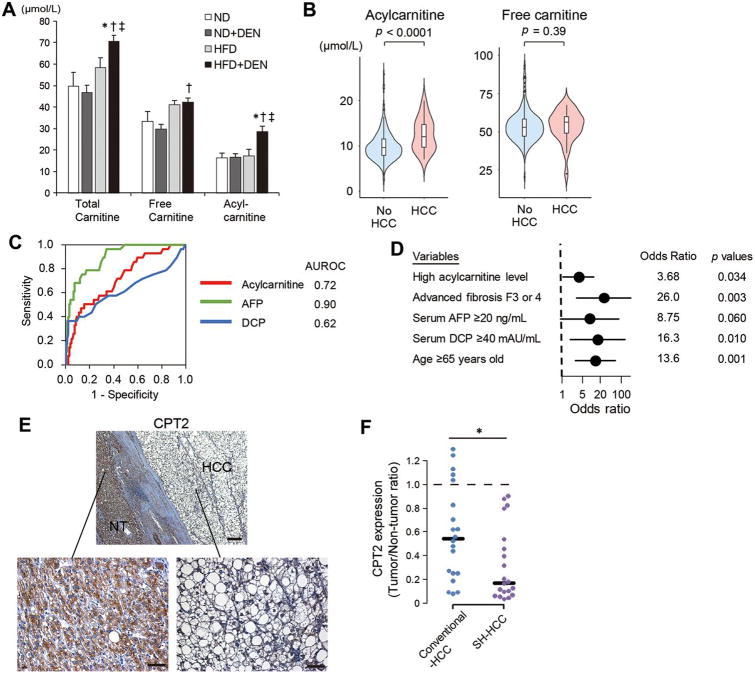
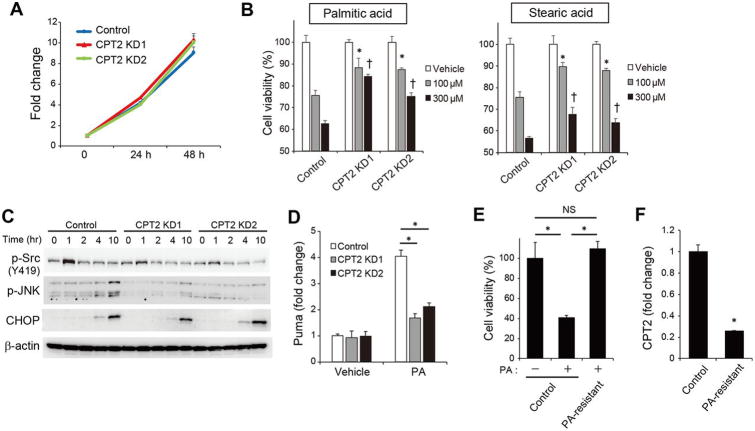
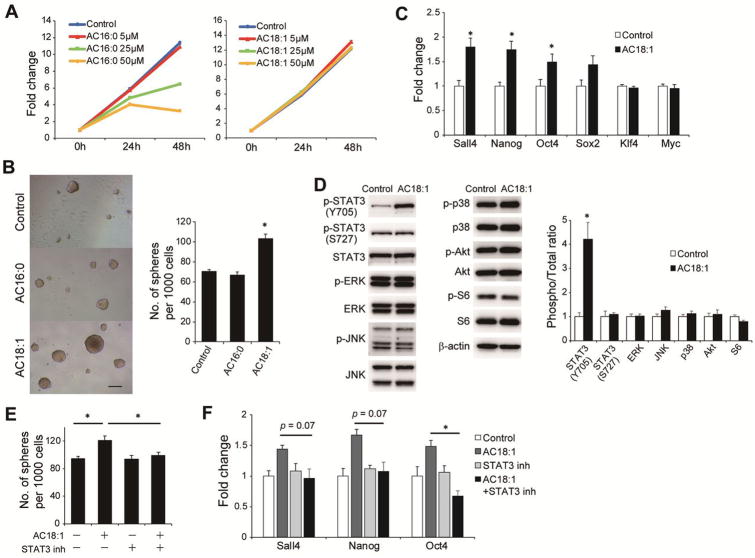
The extensive accumulation of acylcarnitine species was seen in HCC tissues and in the serum of HFD-fed mice. A similar increase was found in the serum of patients with NASH-HCC. The accumulation of acylcarnitine could be attributed to the downregulation of carnitine palmitoyltransferase 2 (CPT2), which was also seen in human SH-HCC. CPT2 downregulation induced the suppression of fatty acid β-oxidation, which would account for the steatotic changes in HCC. CPT2 knockdown in HCC cells resulted in their resistance to lipotoxicity by inhibiting the Src-mediated JNK activation. Additionally, oleoylcarnitine enhanced sphere formation by HCC cells via STAT3 activation, suggesting that acylcarnitine accumulation was a surrogate marker of CPT2 downregulation and directly contributed to hepatocarcinogenesis. HFD feeding and carnitine supplementation synergistically enhanced HCC development accompanied by acylcarnitine accumulation in vivo.
In obesity-driven and NASH-driven HCC, metabolic reprogramming mediated by the downregulation of CPT2 enables HCC cells to escape lipotoxicity and promotes hepatocarcinogenesis.
肿瘤细胞的代谢重编程使其能够适应其局部环境,这是癌症的一个标志。有趣的是,肥胖驱动和非酒精性脂肪性肝炎(NASH)驱动的肝细胞癌(HCC)小鼠模型通常在肿瘤细胞中表现出强烈的脂肪变性,如人类脂肪性肝炎相关 HCC(SH-HCC)中所见,这可能反映了一种特征性的代谢改变。
用二乙基亚硝胺注射的小鼠进行非肿瘤和 HCC 组织的研究,这些小鼠分别喂食正常饮食或高脂肪饮食(HFD),并对其进行全面代谢组学分析,评估肥胖介导的代谢改变在肝癌发生中的意义。
在 HCC 组织和 HFD 喂养小鼠的血清中观察到大量酰基辅酶 A 物质的积累。在 NASH-HCC 患者的血清中也发现了类似的增加。酰基辅酶 A 的积累归因于肉碱棕榈酰转移酶 2(CPT2)的下调,这在人类 SH-HCC 中也观察到。CPT2 的下调诱导了脂肪酸β氧化的抑制,这可以解释 HCC 中的脂肪变性变化。CPT2 在 HCC 细胞中的敲低导致其通过抑制Src 介导的 JNK 激活来抵抗脂毒性。此外,油酰基肉碱通过激活 STAT3 增强 HCC 细胞的球体形成,表明酰基辅酶 A 的积累是 CPT2 下调的替代标志物,并直接促进肝癌发生。HFD 喂养和肉碱补充在体内协同增强 HCC 发展,并伴有酰基辅酶 A 的积累。
在肥胖驱动和 NASH 驱动的 HCC 中,CPT2 下调介导的代谢重编程使 HCC 细胞能够逃避脂毒性,并促进肝癌发生。