Tokarz Rafal, Sameroff Stephen, Tagliafierro Teresa, Jain Komal, Williams Simon H, Cucura D Moses, Rochlin Ilia, Monzon Javier, Carpi Giovanna, Tufts Danielle, Diuk-Wasser Maria, Brinkerhoff Jory, Lipkin W Ian
Center for Infection and Immunity, Mailman School of Public Health, Columbia University, New York, New York, USA.
Division of Vector Control, Suffolk County Department of Public Works, Yaphank, New York, USA.
mSphere. 2018 Mar 7;3(2). doi: 10.1128/mSphere.00614-17. eCollection 2018 Mar-Apr.
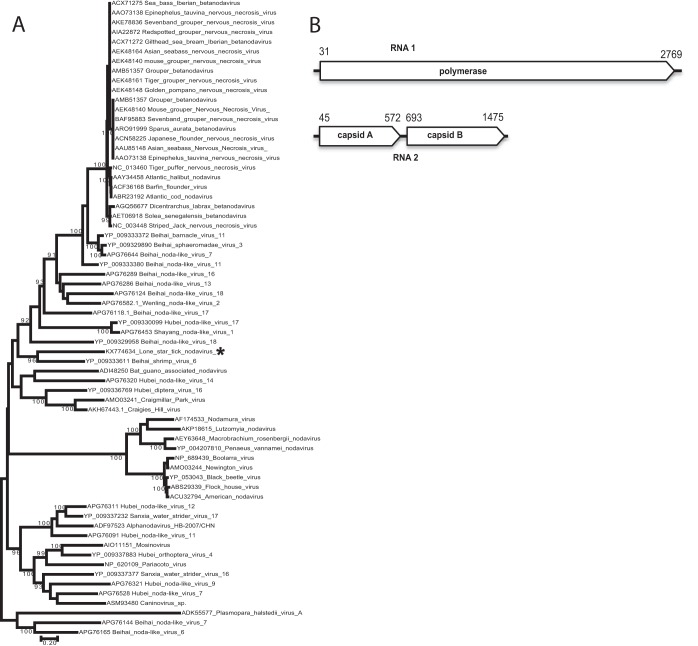
Ticks carry a wide range of known human and animal pathogens and are postulated to carry others with the potential to cause disease. Here we report a discovery effort wherein unbiased high-throughput sequencing was used to characterize the virome of 2,021 ticks, including ( 1,138), ( 720), and ( 163), collected in New York, Connecticut, and Virginia in 2015 and 2016. We identified 33 viruses, including 24 putative novel viral species. The most frequently detected viruses were phylogenetically related to members of the and families, as well as the recently proposed . Our work expands our understanding of tick viromes and underscores the high viral diversity that is present in ticks. The incidence of tick-borne disease is increasing, driven by rapid geographical expansion of ticks and the discovery of new tick-associated pathogens. The examination of the tick microbiome is essential in order to understand the relationship between microbes and their tick hosts and to facilitate the identification of new tick-borne pathogens. Genomic analyses using unbiased high-throughput sequencing platforms have proven valuable for investigations of tick bacterial diversity, but the examination of tick viromes has historically not been well explored. By performing a comprehensive virome analysis of the three primary tick species associated with human disease in the United States, we gained substantial insight into tick virome diversity and can begin to assess a potential role of these viruses in the tick life cycle.
蜱虫携带多种已知的人类和动物病原体,据推测还携带其他可能致病的病原体。在此,我们报告一项发现工作,其中利用无偏差高通量测序对2015年和2016年在纽约、康涅狄格州和弗吉尼亚州采集的2021只蜱虫(包括1138只肩突硬蜱、720只篦子硬蜱和163只美洲钝缘蜱)的病毒组进行了特征描述。我们鉴定出33种病毒,包括24种假定的新型病毒物种。检测到的最常见病毒在系统发育上与呼肠孤病毒科和弹状病毒科的成员以及最近提出的Megabirnaviridae科相关。我们的工作扩展了我们对蜱虫病毒组的认识,并强调了蜱虫中存在的高度病毒多样性。蜱传疾病的发病率正在上升,这是由蜱虫的快速地理扩张和新的蜱虫相关病原体的发现所驱动的。对蜱虫微生物组的检查对于理解微生物与其蜱虫宿主之间的关系以及促进新的蜱传病原体的鉴定至关重要。使用无偏差高通量测序平台进行的基因组分析已被证明对蜱虫细菌多样性的研究很有价值,但蜱虫病毒组的检查在历史上并未得到充分探索。通过对与美国人类疾病相关的三种主要蜱虫物种进行全面的病毒组分析,我们对蜱虫病毒组多样性有了深入了解,并可以开始评估这些病毒在蜱虫生命周期中的潜在作用。