Guo Junhui, Zhao Yun, Jiang Xingpeng, Li Rui, Xie Hao, Ge Leixin, Xie Bo, Yang Xu, Zhang Luoping
Hubei Key Laboratory of Genetic Regulation and Integrative Biology, School of Life Sciences, Central China Normal University, Wuhan 430079, China.
Division of Environmental Health Sciences, School of Public Health, University of California, Berkeley, CA 94720, USA.
Genes (Basel). 2018 Apr 3;9(4):192. doi: 10.3390/genes9040192.
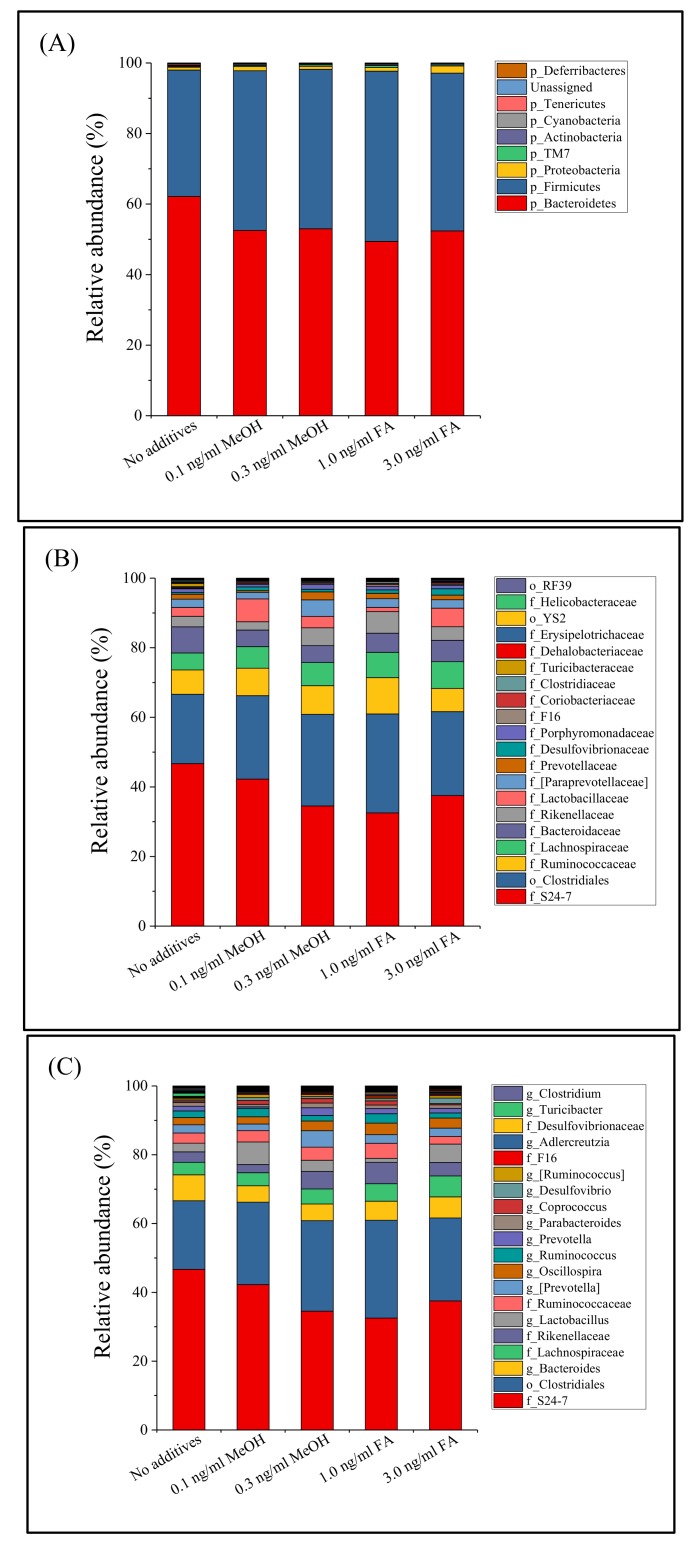
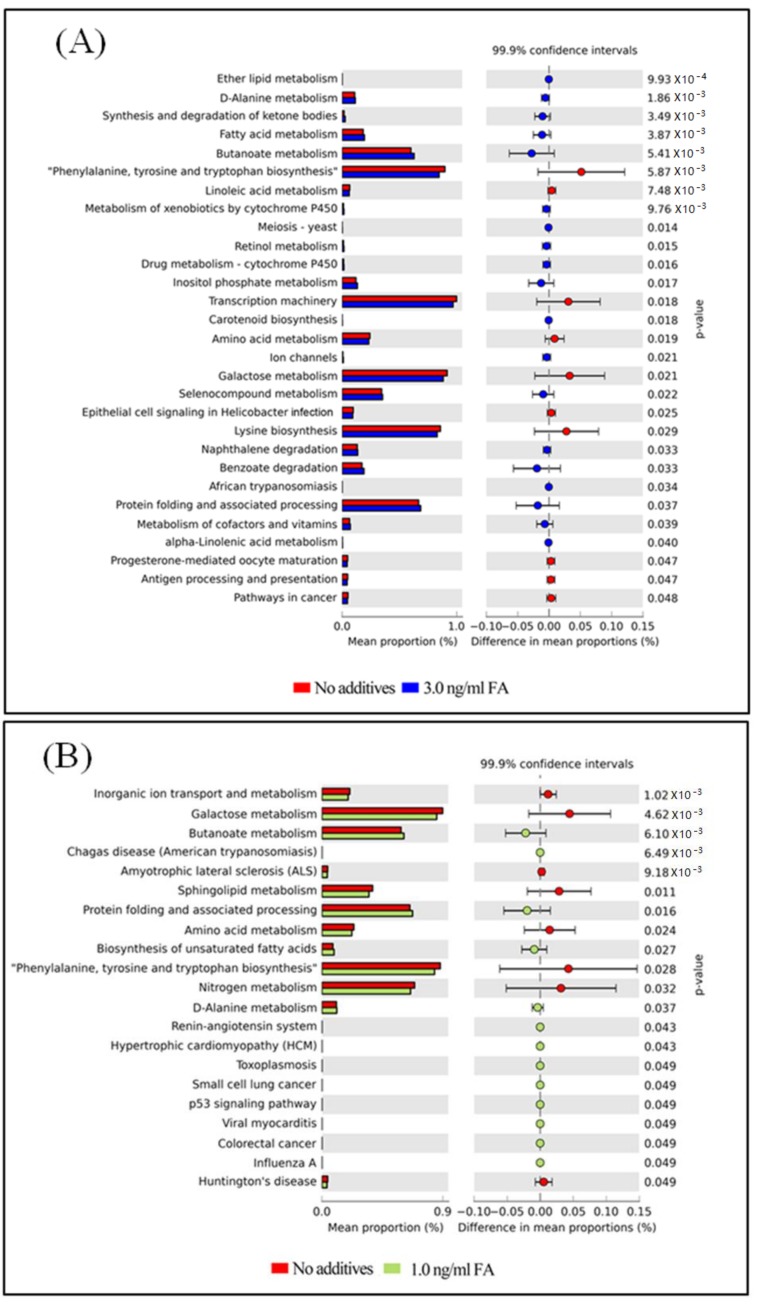
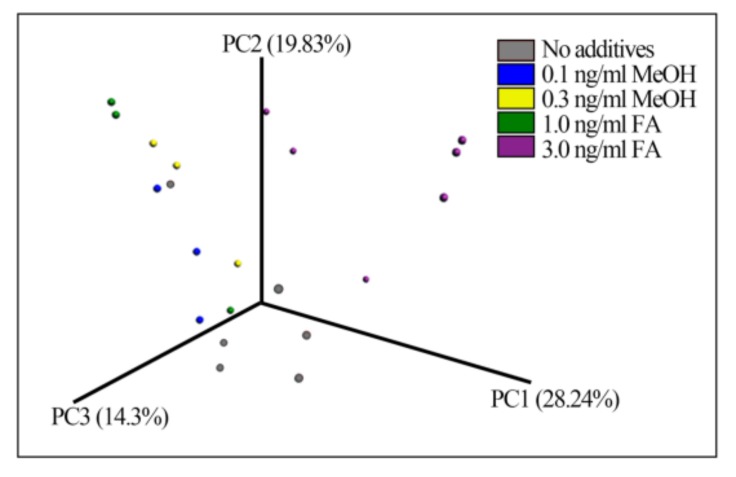
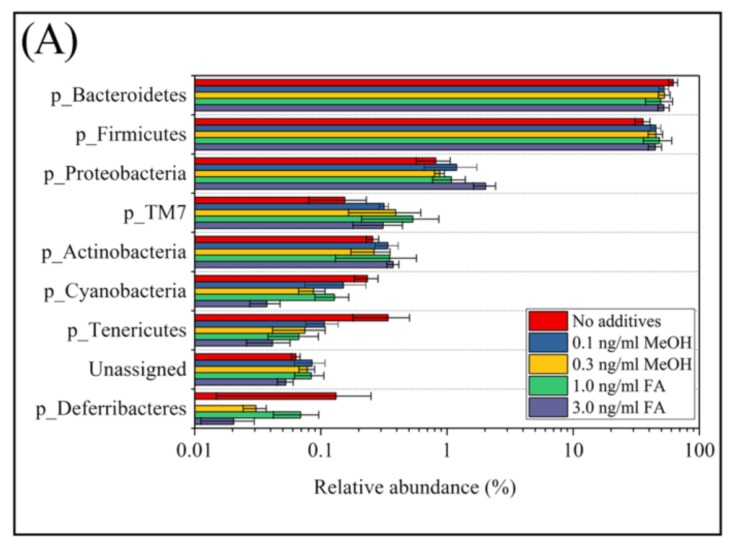
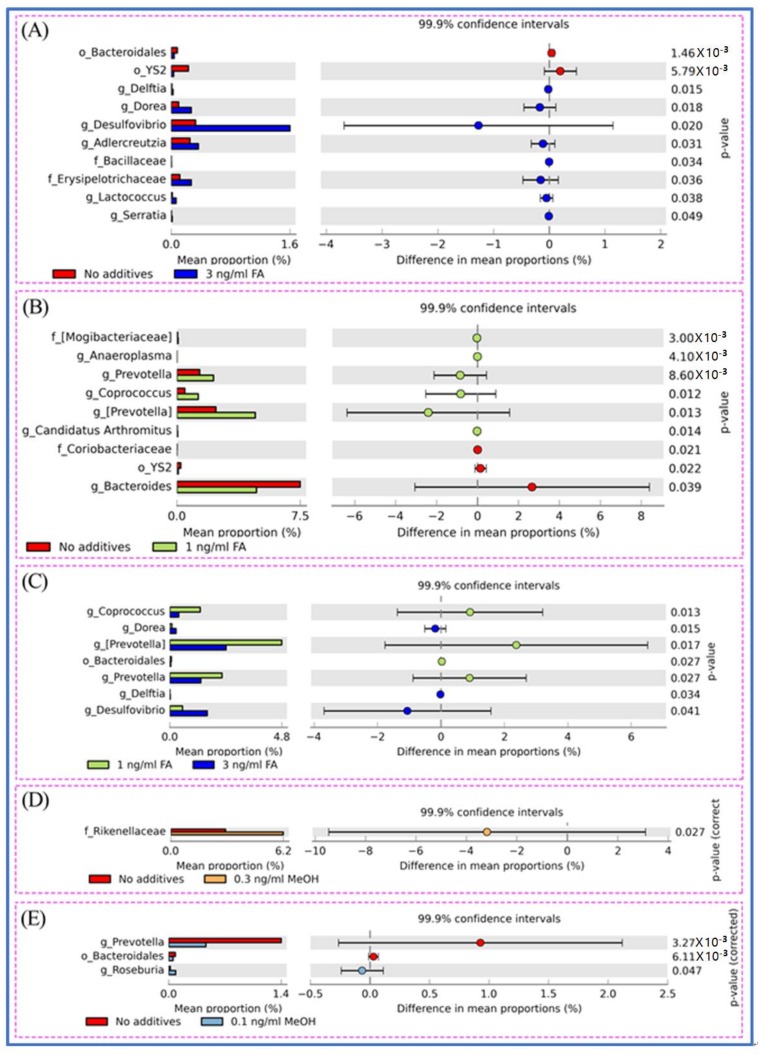
Exposure to Formaldehyde (FA) results in many pathophysiological symptoms, however the underlying mechanisms are not well understood. Given the complicated modulatory role of intestinal microbiota on human health, we hypothesized that interactions between FA and the gut microbiome may account for FA's toxicity. Balb/c mice were allocated randomly to three groups: a control group, a methanol group (0.1 and 0.3 ng/mL MeOH subgroups), and an FA group (1 and 3 ng/mL FA subgroups). Groups of either three or six mice were used for the control or experiment. We applied high-throughput sequencing of 16S ribosomal RNA (rRNA) gene approaches and investigated possible alterations in the composition of mouse gut microbiota induced by FA. Changes in bacterial genera induced by FA exposure were identified. By analyzing KEGG metabolic pathways predicted by PICRUSt software, we also explored the potential metabolic changes, such as alpha-Linolenic acid metabolism and pathways in cancer, associated with FA exposure in mice. To the best of our knowledge, this preliminary study is the first to identify changes in the mouse gut microbiome after FA exposure, and to analyze the relevant potential metabolisms. The limitation of this study: this study is relatively small and needs to be further confirmed through a larger study.
接触甲醛(FA)会导致许多病理生理症状,但其潜在机制尚不清楚。鉴于肠道微生物群对人类健康具有复杂的调节作用,我们推测FA与肠道微生物群之间的相互作用可能是FA毒性的原因。将Balb/c小鼠随机分为三组:对照组、甲醇组(0.1和0.3 ng/mL甲醇亚组)和FA组(1和3 ng/mL FA亚组)。每组三只或六只小鼠用于对照或实验。我们应用16S核糖体RNA(rRNA)基因高通量测序方法,研究FA诱导的小鼠肠道微生物群组成的可能变化。确定了FA暴露诱导的细菌属变化。通过分析PICRUSt软件预测的KEGG代谢途径,我们还探索了与小鼠FA暴露相关的潜在代谢变化,如α-亚麻酸代谢和癌症相关途径。据我们所知,这项初步研究首次确定了FA暴露后小鼠肠道微生物群的变化,并分析了相关的潜在代谢。本研究的局限性:本研究规模相对较小,需要通过更大规模的研究进一步证实。