Computational Biology Unit, Research and Innovation Centre, Fondazione Edmund Mach, via E. Mach 1, 38010 S. Michele all'Adige (TN), Italy.
Gigascience. 2018 Apr 1;7(4):1-8. doi: 10.1093/gigascience/giy032.
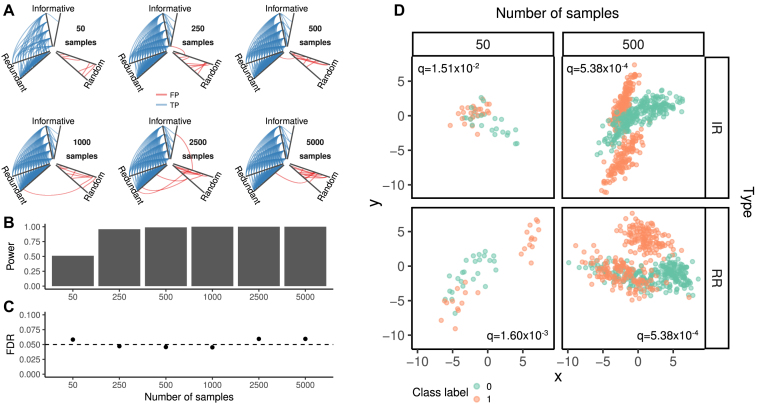
The ability of finding complex associations in large omics datasets, assessing their significance, and prioritizing them according to their strength can be of great help in the data exploration phase. Mutual information-based measures of association are particularly promising, in particular after the recent introduction of the TICe and MICe estimators, which combine computational efficiency with superior bias/variance properties. An open-source software implementation of these two measures providing a complete procedure to test their significance would be extremely useful.
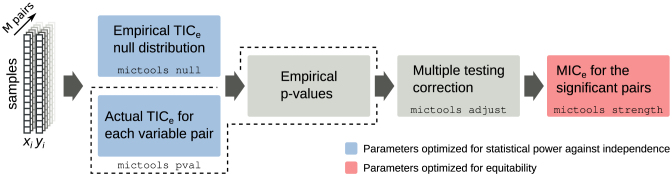
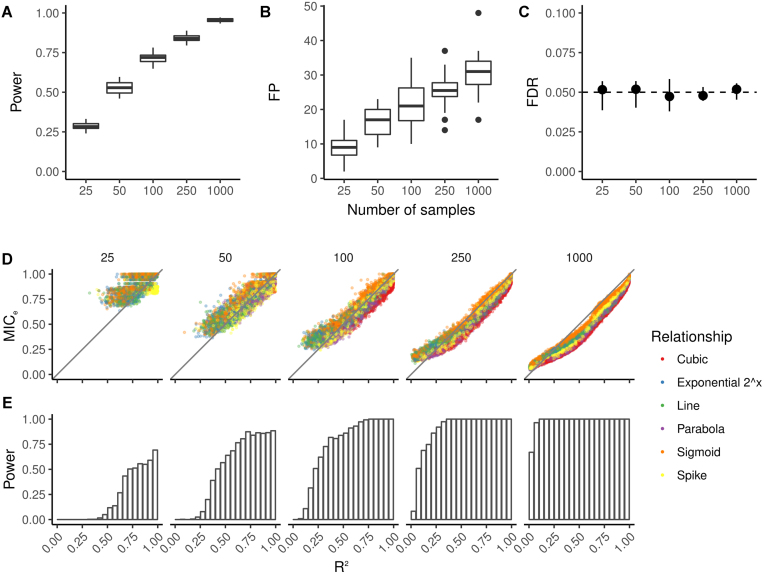
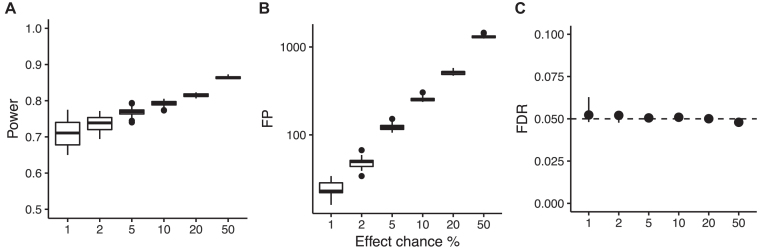
Here, we present MICtools, a comprehensive and effective pipeline that combines TICe and MICe into a multistep procedure that allows the identification of relationships of various degrees of complexity. MICtools calculates their strength assessing statistical significance using a permutation-based strategy. The performances of the proposed approach are assessed by an extensive investigation in synthetic datasets and an example of a potential application on a metagenomic dataset is also illustrated.
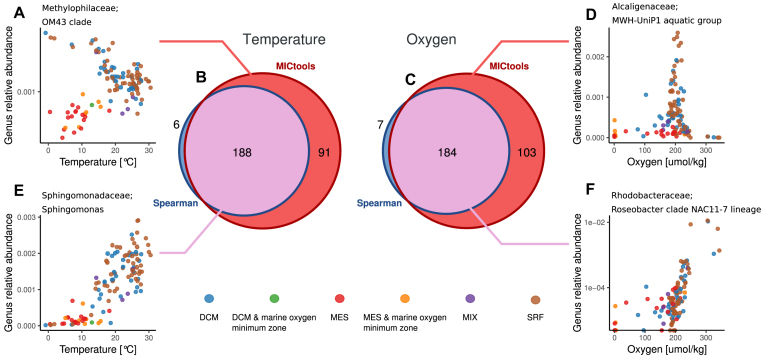
We show that MICtools, combining TICe and MICe, is able to highlight associations that would not be captured by conventional strategies.
在数据探索阶段,发现大型组学数据集中复杂关联的能力、评估其显著性并根据其强度对其进行优先级排序,可能会有很大帮助。基于互信息的关联度量方法特别有前途,特别是在最近引入了 TICe 和 MICe 估计量之后,它们将计算效率与优越的偏差/方差特性结合在一起。一个开源软件实现这两种度量标准,并提供一个完整的程序来测试它们的显著性,将是非常有用的。
在这里,我们提出了 MICtools,这是一个全面有效的管道,将 TICe 和 MICe 结合到一个多步骤的过程中,该过程允许识别各种复杂程度的关系。MICtools 使用基于排列的策略来计算它们的强度,并评估统计显著性。通过在合成数据集上的广泛调查和对宏基因组数据集的潜在应用示例,评估了所提出方法的性能。
我们表明,MICtools 将 TICe 和 MICe 结合在一起,能够突出传统策略无法捕捉到的关联。