Yinda Claude Kwe, Ghogomu Stephen Mbigha, Conceição-Neto Nádia, Beller Leen, Deboutte Ward, Vanhulle Emiel, Maes Piet, Van Ranst Marc, Matthijnssens Jelle
Laboratory of Viral Metagenomics.
Laboratory for Clinical and Epidemiological Virology, Department of Microbiology and Immunology, Rega Institute, KU Leuven - University of Leuven, B-3000 Leuven, Belgium.
Virus Evol. 2018 Mar 30;4(1):vey008. doi: 10.1093/ve/vey008. eCollection 2018 Jan.
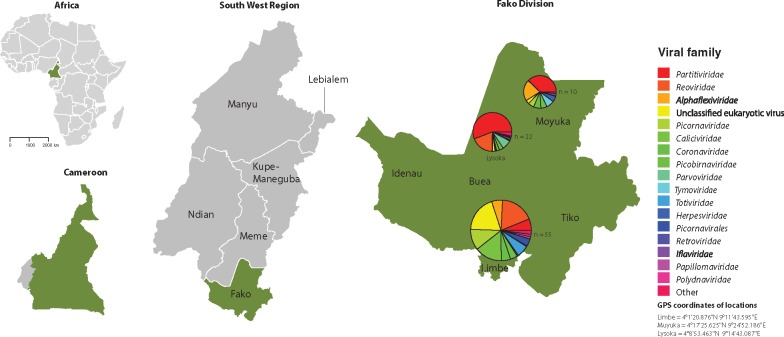
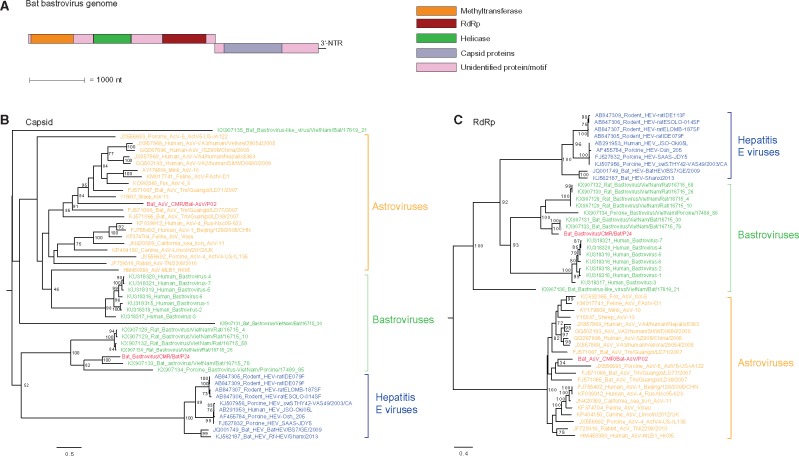
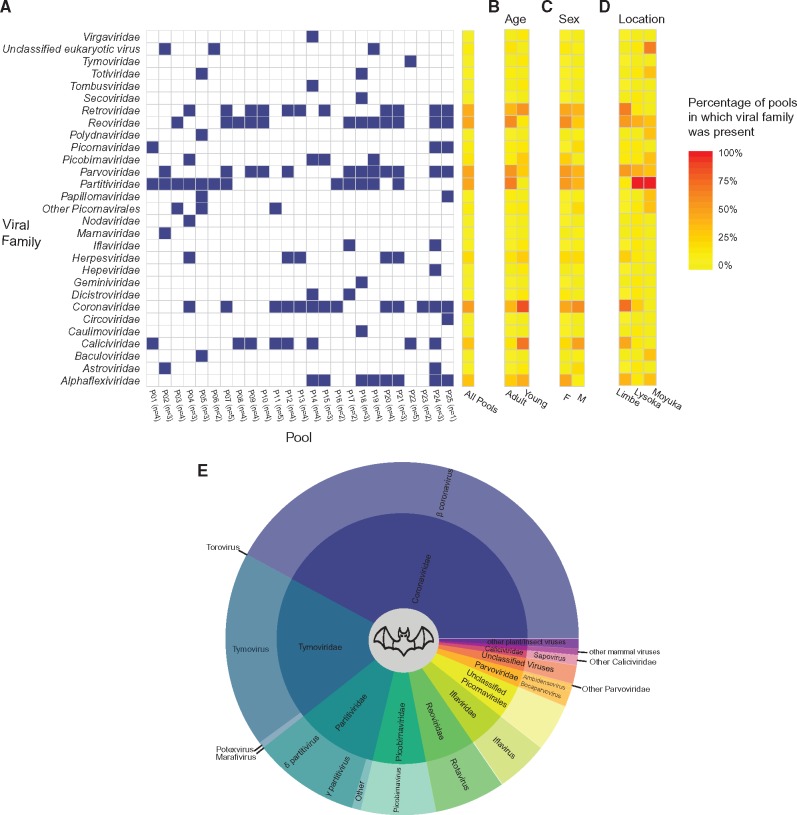
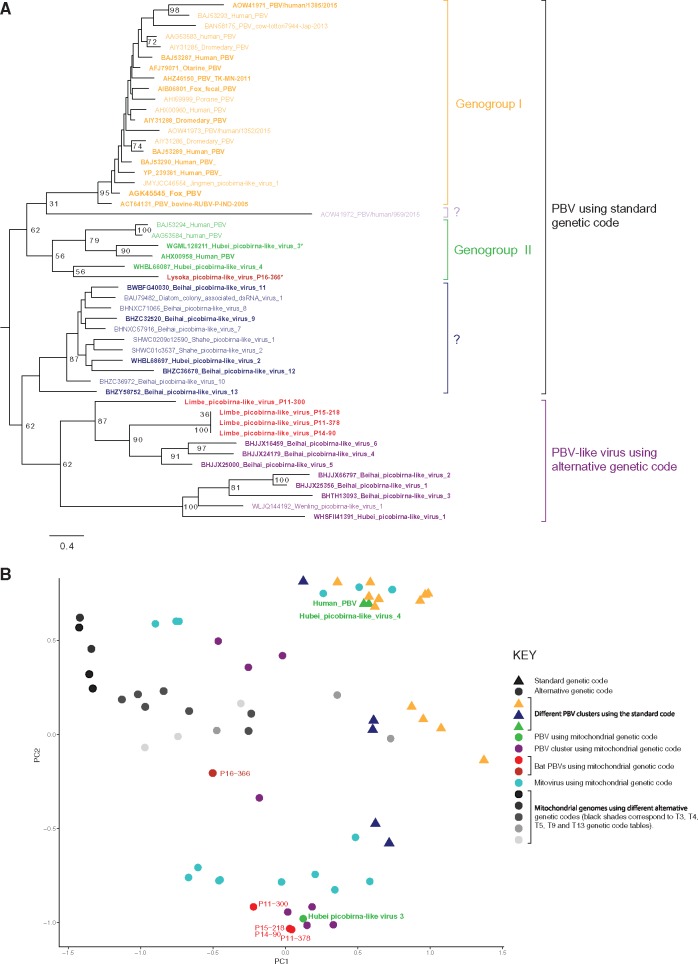
Most human emerging infectious diseases originate from wildlife and bats are a major reservoir of viruses, a few of which have been highly pathogenic to humans. In some regions of Cameroon, bats are hunted and eaten as a delicacy. This close proximity between human and bats provides ample opportunity for zoonotic events. To elucidate the viral diversity of Cameroonian fruit bats, we collected and metagenomically screened eighty-seven fecal samples of and fruit bats. The results showed a plethora of known and novel viruses. Phylogenetic analyses of the eleven gene segments of the first complete bat rotavirus H genome, showed clearly separated clusters of human, porcine, and bat rotavirus H strains, not indicating any recent interspecies transmission events. Additionally, we identified and analyzed a bat bastrovirus genome (a novel group of recently described viruses, related to astroviruses and hepatitis E viruses), confirming their recombinant nature, and provide further evidence of additional recombination events among bat bastroviruses. Interestingly, picobirnavirus-like RNA-dependent RNA polymerase gene segments were identified using an alternative mitochondrial genetic code, and further principal component analyses suggested that they may have a similar lifestyle to mitoviruses, a group of virus-like elements known to infect the mitochondria of fungi. Although identified bat coronavirus, parvovirus, and cyclovirus strains belong to established genera, most of the identified partitiviruses and densoviruses constitute putative novel genera in their respective families. Finally, the results of the phage community analyses of these bats indicate a very diverse geographically distinct bat phage population, probably reflecting different diets and gut bacterial ecosystems.
大多数人类新发传染病起源于野生动物,而蝙蝠是病毒的主要宿主,其中一些病毒对人类具有高度致病性。在喀麦隆的一些地区,蝙蝠被作为美味捕杀食用。人类与蝙蝠的这种密切接触为动物源性疾病的发生提供了充足机会。为了阐明喀麦隆果蝠的病毒多样性,我们收集并通过宏基因组学方法筛选了87份锤头果蝠和 Franquet's epauletted 果蝠的粪便样本。结果显示存在大量已知和新型病毒。对首个完整的蝙蝠轮状病毒H基因组的11个基因片段进行系统发育分析,结果表明人、猪和蝙蝠轮状病毒H毒株形成明显分离的簇,未显示近期有任何种间传播事件。此外,我们鉴定并分析了一种蝙蝠杆状套病毒基因组(一组最近描述的新型病毒,与星状病毒和戊型肝炎病毒相关),证实了它们的重组性质,并为蝙蝠杆状套病毒之间的其他重组事件提供了进一步证据。有趣的是,类微小双股RNA病毒样RNA依赖RNA聚合酶基因片段是使用一种替代的线粒体遗传密码鉴定出来的,进一步的主成分分析表明它们可能具有与线粒体病毒类似的生活方式,线粒体病毒是已知感染真菌线粒体的一类病毒样元件。虽然鉴定出的蝙蝠冠状病毒、细小病毒和环病毒毒株属于已确定的属,但大多数鉴定出的双链RNA病毒和浓病毒在各自家族中构成推定的新属。最后,对这些蝙蝠的噬菌体群落分析结果表明,蝙蝠噬菌体群体在地理上非常多样化,可能反映了不同的饮食和肠道细菌生态系统。