Kim Yeonjoon, Kim Jin Woo, Kim Zeehyo, Kim Woo Youn
Department of Chemistry , KAIST , 291 Daehak-ro, Yuseong-gu , Daejeon 34141 , Korea . Email:
Chem Sci. 2017 Dec 12;9(4):825-835. doi: 10.1039/c7sc03628k. eCollection 2018 Jan 28.
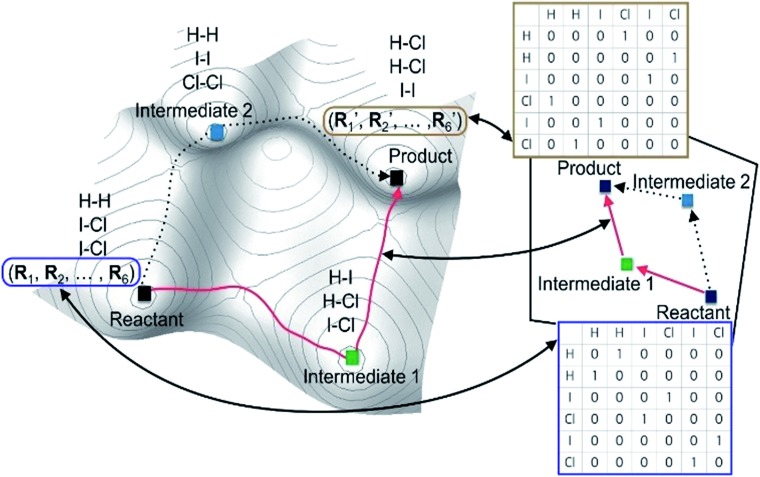
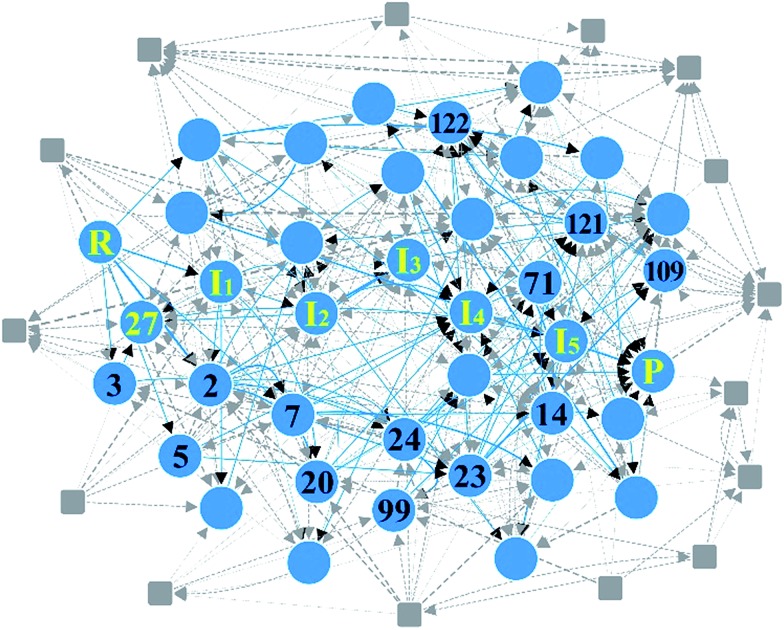
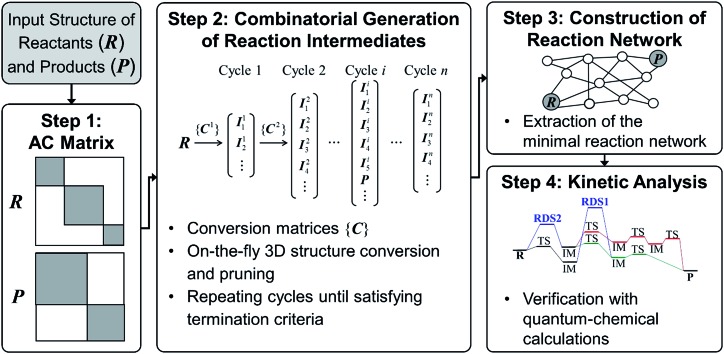
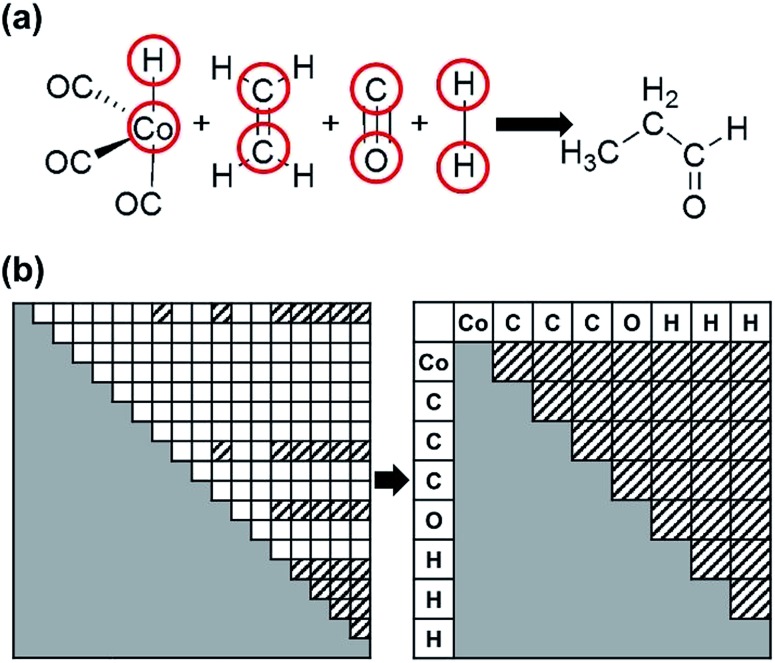
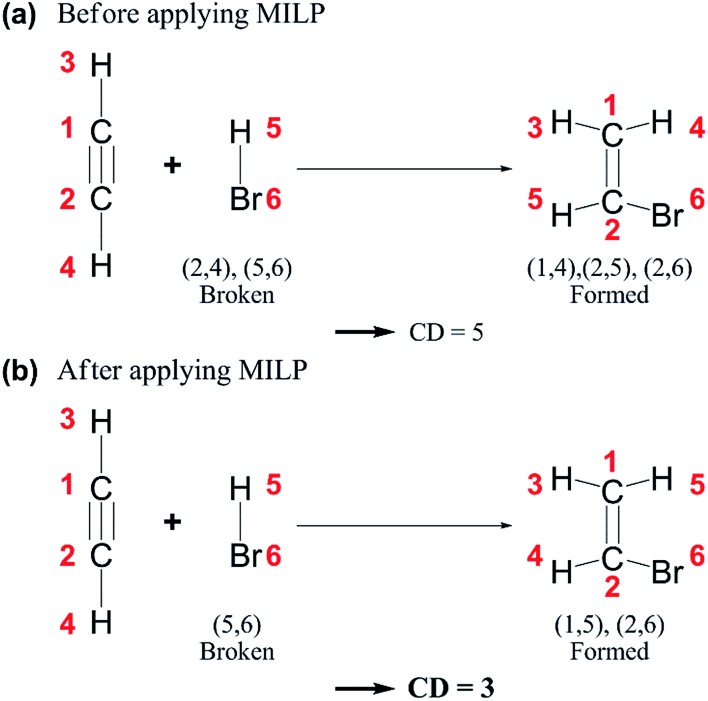
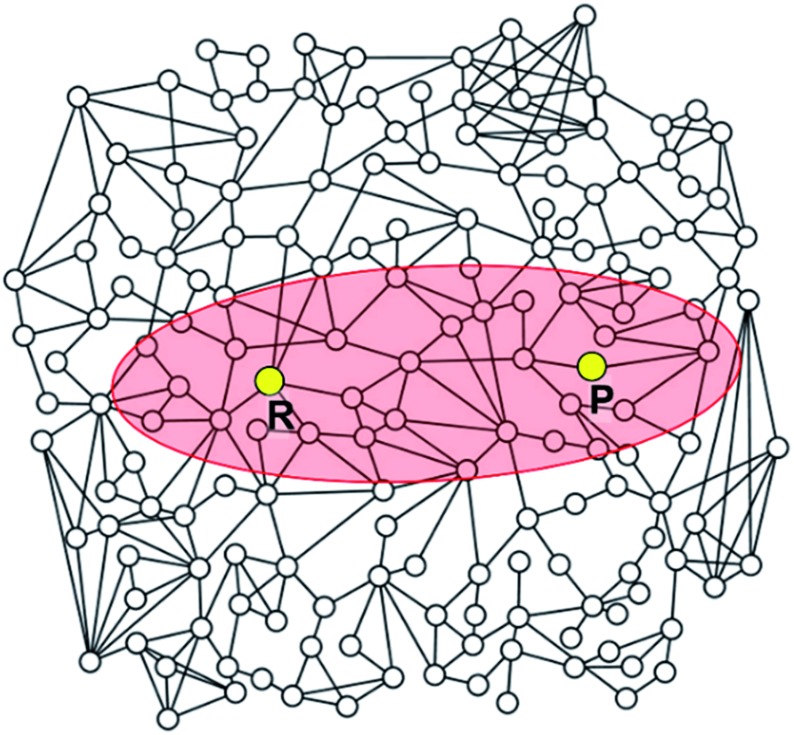
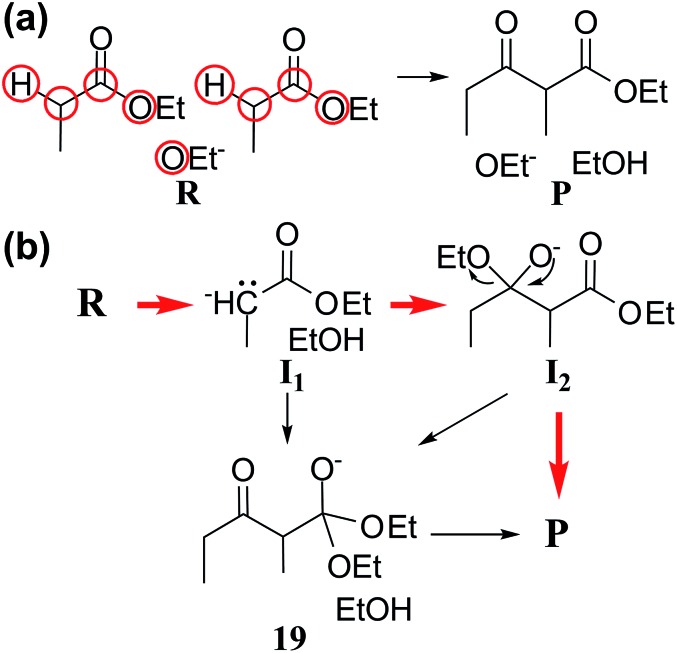
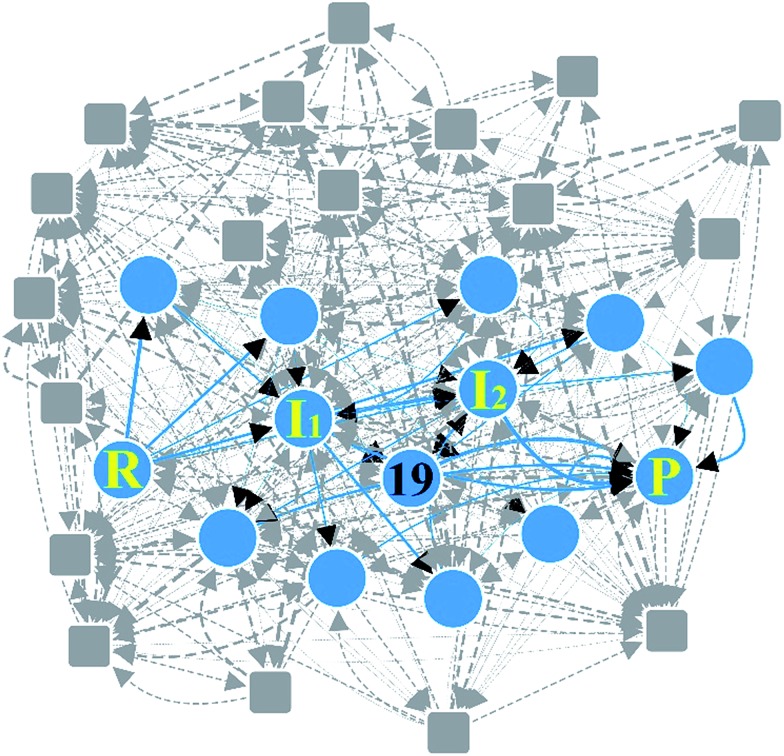
Despite remarkable advances in computational chemistry, prediction of reaction mechanisms is still challenging, because investigating all possible reaction pathways is computationally prohibitive due to the high complexity of chemical space. A feasible strategy for efficient prediction is to utilize chemical heuristics. Here, we propose a novel approach to rapidly search reaction paths in a fully automated fashion by combining chemical theory and heuristics. A key idea of our method is to extract a minimal reaction network composed of only favorable reaction pathways from the complex chemical space through molecular graph and reaction network analysis. This can be done very efficiently by exploring the routes connecting reactants and products with minimum dissociation and formation of bonds. Finally, the resulting minimal network is subjected to quantum chemical calculations to determine kinetically the most favorable reaction path at the predictable accuracy. As example studies, our method was able to successfully find the accepted mechanisms of Claisen ester condensation and cobalt-catalyzed hydroformylation reactions.
尽管计算化学取得了显著进展,但反应机理的预测仍然具有挑战性,因为由于化学空间的高度复杂性,研究所有可能的反应途径在计算上是 prohibitive 的。一种可行的高效预测策略是利用化学启发法。在这里,我们提出了一种新颖的方法,通过结合化学理论和启发法以全自动方式快速搜索反应路径。我们方法的一个关键思想是通过分子图和反应网络分析从复杂的化学空间中提取仅由有利反应途径组成的最小反应网络。这可以通过探索连接反应物和产物且键的解离和形成最少的路线非常有效地完成。最后,对所得的最小网络进行量子化学计算,以在可预测的精度下从动力学上确定最有利的反应路径。作为实例研究,我们的方法能够成功找到克莱森酯缩合反应和钴催化的氢甲酰化反应的公认机理。