Department of Computer Science, Texas State University, San Marcos, Texas, 78666, USA.
Michael Smith Genome Sciences Centre, British Columbia Cancer Agency, Vancouver, British Columbia, V5Z 1L3, Canada.
Sci Rep. 2018 May 3;8(1):6951. doi: 10.1038/s41598-018-24758-5.
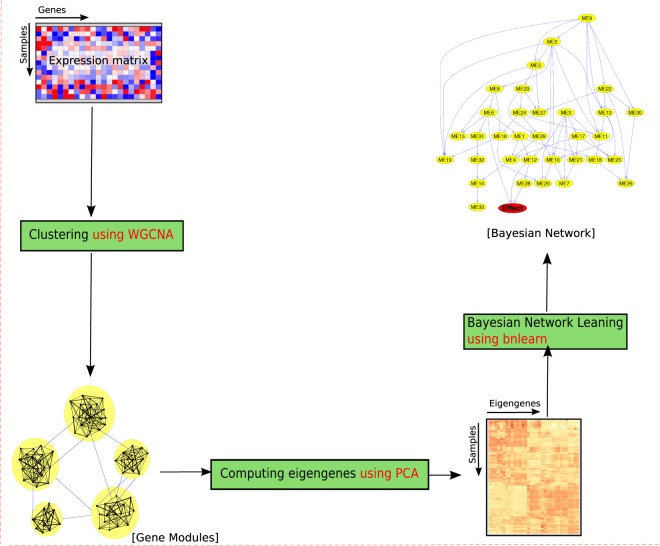
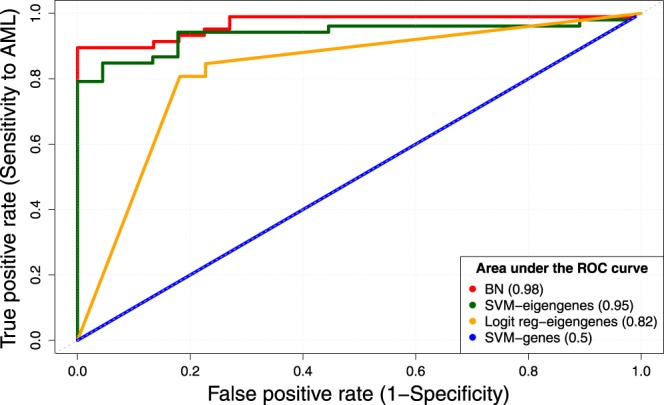
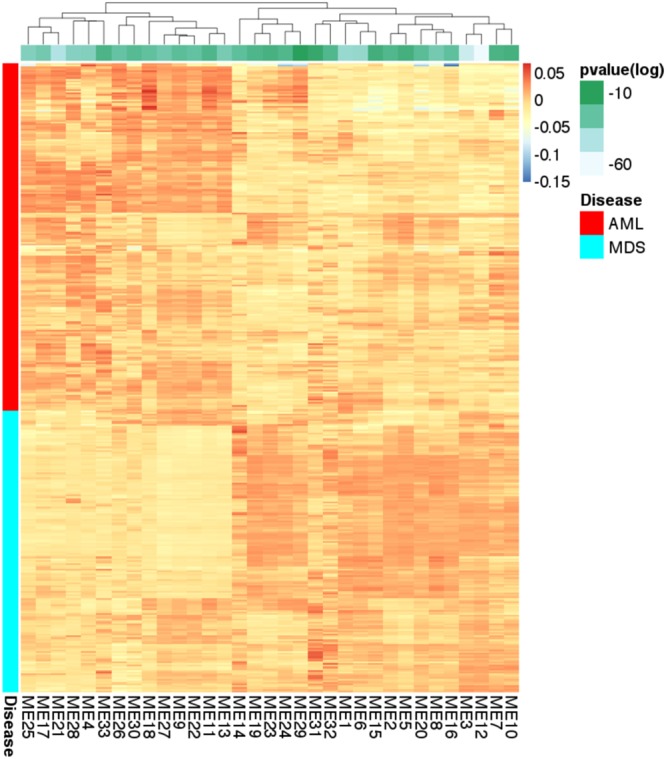
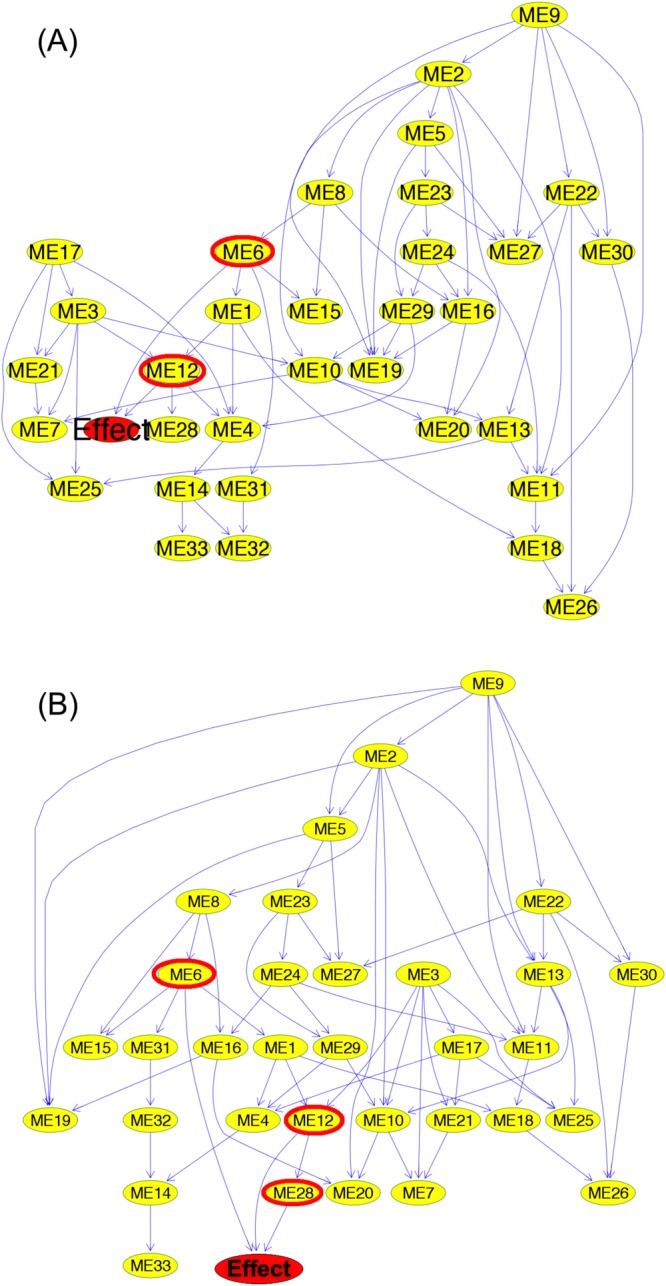
Network analysis is the preferred approach for the detection of subtle but coordinated changes in expression of an interacting and related set of genes. We introduce a novel method based on the analyses of coexpression networks and Bayesian networks, and we use this new method to classify two types of hematological malignancies; namely, acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). Our classifier has an accuracy of 93%, a precision of 98%, and a recall of 90% on the training dataset (n = 366); which outperforms the results reported by other scholars on the same dataset. Although our training dataset consists of microarray data, our model has a remarkable performance on the RNA-Seq test dataset (n = 74, accuracy = 89%, precision = 88%, recall = 98%), which confirms that eigengenes are robust with respect to expression profiling technology. These signatures are useful in classification and correctly predicting the diagnosis. They might also provide valuable information about the underlying biology of diseases. Our network analysis approach is generalizable and can be useful for classifying other diseases based on gene expression profiles. Our previously published Pigengene package is publicly available through Bioconductor, which can be used to conveniently fit a Bayesian network to gene expression data.
网络分析是检测相互作用和相关基因集表达细微但协调变化的首选方法。我们引入了一种基于共表达网络和贝叶斯网络分析的新方法,并使用这种新方法对两种血液系统恶性肿瘤(即急性髓系白血病(AML)和骨髓增生异常综合征(MDS))进行分类。我们的分类器在训练数据集(n=366)上的准确率为 93%,精度为 98%,召回率为 90%;优于其他学者在同一数据集上报告的结果。虽然我们的训练数据集包含微阵列数据,但我们的模型在 RNA-Seq 测试数据集(n=74)上表现出色(准确率=89%,精度=88%,召回率=98%),这证实了特征基因在表达谱技术方面是稳健的。这些特征在分类和正确预测诊断方面非常有用。它们还可能提供有关疾病潜在生物学的有价值信息。我们的网络分析方法具有通用性,可用于根据基因表达谱对其他疾病进行分类。我们之前发布的 Pigengene 软件包可通过 Bioconductor 公开获得,可用于方便地将贝叶斯网络拟合到基因表达数据。